Complement-system.eu
The American Journal of Pathology, Vol. 176, No. 4, April 2010
Copyright American Society for Investigative Pathology
Cardiovascular, Pulmonary and Renal Pathology
Therapeutic Targeting of Classical and LectinPathways of Complement Protects fromIschemia-Reperfusion-Induced Renal Damage
Giuseppe Castellano,* Rita Melchiorre,*
of classical and lectin pathways of complement in a
Antonia Loverre,* Pasquale Ditonno,†
swine model of ischemia-reperfusionⴚ
induced renal
Vincenzo Montinaro,* Michele Rossini,*
damage. Therefore , inhibition of these two pathways
Chiara Divella,* Michele Battaglia,†
might represent a novel therapeutic approach in the
Giuseppe Lucarelli,† Gennaro Annunziata,†
prevention of delayed graft function in kidney trans-
plant recipients.
Silvano Palazzo,† Francesco Paolo Selvaggi,†
(Am J Pathol 2010, 176:1648 –1659; DOI:
Francesco Staffieri,‡ Antonio Crovace,‡Mohamed R. Daha,§ Maurice Mannesse,¶Sandra van Wetering,¶ Francesco Paolo Schena,*
Delayed graft function (DGF) is the primary early post-
and Giuseppe Grandaliano*
transplant complication of kidney recipients.1 This event,
From the Renal, Dialysis and Transplantation Unit,*
Urology,
histologically characterized by the presence of acute tubular
Andrology and Renal Transplantation Unit,†
Veterinary Surgery Unit,‡
necrosis, has been reported to occur in 25% to 30% of renal
and the Department of Emergency and Organ Transplantation,
transplants.1 DGF is commonly associated with a significantly
University of Bari, Bari, Italy; the Department of Nephrology,§
Leiden
longer hospital stay and an increase in peritransplant morbid-
University Medical Center, Leiden; The Netherlands, and Research
ity. Moreover, this early complication seems to result in a
Laboratories,¶
Pharming Group NV, Leiden, The Netherlands
marked reduction in long-term graft survival.2 Indeed, DGF isassociated with an increased rate of acute rejection and asuboptimal renal function at 1-year post-transplantation.1–3
Ischemia followed by reperfusion plays a pivotal role in
the pathogenesis of early graft damage.1 Ischemia-
Ischemia-reperfusion injury is the major cause of de-
reperfusion injury is characterized by two main features
layed graft function in transplanted kidneys , an early
at the renal level: apoptosis of tubular cells and interstitial
event significantly affecting long-term graft function
and survival. Several studies in rodents suggest that
inflammation. Although many steps in the cascade of
the alternative pathway of the complement system
events leading to ischemia-reperfusion injury are unclear,
plays a pivotal role in renal ischemia-reperfusion in-
the most promising potential mechanisms include recruit-
jury. However , limited information is currently avail-
ment and activation of inflammatory cells and local prim-
able from humans and larger animals. Here we dem-
ing of the complement cascade.4 – 6
onstrated that 30 minutes of ischemia resulted in the
The complement system is a major constituent of the
induction of C4d/C1q , C4d/MLB , and MBL/MASP-2 de-
innate immune system, participating in the pathogenesis
posits in a swine model of ischemia-reperfusion in-
jury. The infusion of C1-inhibitor led to a significant
Supported by University of Bari and Regione Puglia (PhD in Biotechnology
reduction in peritubular capillary and glomerular C4d
applied to Organ and Tissue Transplantation, R.M. and C.D.) and an unre-
and C5b-9 deposition. Moreover , complement-inhib-
stricted research grant from Pharming Group NV, Leiden, The Netherlands.
iting treatment significantly reduced the numbers
G.C. and R.M. equally contributed to the present study.
of infiltrating CD163ⴙ
, SWC3aⴙ
, CD4aⴙ
, and CD8aⴙ
Accepted for publication December 4, 2009.
cells. C1-inhibitor administration led to significant
Pharming Group NV has a financial interest in the use of recombinant
inhibition of tubular damage and tubular epithelial
human C1 inhibitor (rhC1NH) in preventing delayed graft function after
cells apoptosis. Interestingly , we report that focal
solid organ transplantation.
C4d-deposition colocalizes with C1q and MBL at the
Address reprint requests to Giuseppe Castellano, M.D., Ph.D.; Renal,
peritubular and glomerular capillary levels also in
Dialysis, and Transplantation Unit; Department of Emergency and Organ
patients with delayed graft function. In conclusion ,
Transplantation; University of Bari, Policlinico, Piazza Giulio Cesare 11,
we demonstrated the activation and a pathogenic role
70124 Bari, Italy. E-mail:
[email protected].
C1-Inhibitor in Renal I/R Injury
AJP April 2010, Vol. 176, No. 4
of tissue damage through sequential activation of differ-
formed at 15, 30, and 60 minutes after reperfusion; animals
ent proteases. The priming of this proteolytic cascade
where sacrificed 24 hours after the surgical procedure. A
may occur by classical, alternative, and lectin-mediated
portion of each biopsy specimen was immediately snap-
activation pathways, and generates several pro-inflam-
frozen in optimal cutting temperature (Tissuetek) medium
matory mediators.6,7 Studies in animal models have
and stored in liquid nitrogen. Another portion was fixed in
shown complement activation in the kidney after ischemia-
buffered formalin (4%) for 12 hours and embedded in par-
reperfusion, leading to the generation of several mediators
affin using standard procedures.
of inflammation, such as C3a, C5a, and C5b-9.4 Mice de-ficient in complement components such as C6 show verylimited damage after renal ischemia-reperfusion injury.8
In Vitro
and ex Vivo
Effects of rhC1INH on
Moreover, the use of anti-factor B or C5a-receptor
Swine Complement System
antagonists has been shown to reduce renal damagedue to ischemia-reperfusion.9,10 Therefore, prevention
Sera from three healthy pigs were incubated with
of complement activation is currently considered one
rhC1INH at different concentration (1, 2, 3U/ml) or vehicle
of the best therapeutic targets to prevent or limit isch-
for 30 minutes at 37°C. The inhibition of complement
emia-reperfusion⫺induced renal damage.5,6,11 C1-inhib-
functional activity in classical, alternative, and lectin path-
itor (C1INH) is a potent inhibitor of proteases of the clas-
ways was assessed using a commercially available en-
sical and lectin complement pathways (C1r, C1s, and
zyme immunoassay kit (Wielisa, Wieslab, Lund, Swe-
MASP2).6,11,12 Animal studies show that C1INH can pro-
den).15 Briefly, the assay used a specific coating for each
tect liver, intestine, heart, and brain tissue from ischemia-
pathway so that only activation of that particular pathway
reperfusion damage.13 There are no published data on
occurs up to C9 (human IgM for the classical pathway,
the effect of C1INH on ischemia-reperfusion⫺induced
LPS for the alternative pathway and mannan for MBL
renal damage, since most of the existing evidence sug-
pathway). Complement activity via each pathway was
gests that in this setting complement activation is mainly
detected using a specific monoclonal antibody against
induced through the alternative pathway.5,14 However, all
the C5b-9 complex.
data reported in the literature are from murine models,
In addition, blood samples were collected from three
and no information is currently available from larger ani-
pigs, weighting 20 to 38 kg, at different times (0, 15, 60,
mals and/or human subjects.
and 240 minutes) after the administration of rhC1INH
The aim of the present study was to investigate the
(500 U/kg) to assess complement activation (Wielisa).
pattern of complement activation in patients with DGFand to test the efficacy of a recombinant form of human
C1INH (rhC1INH) in preventing renal damage in a swinemodel of warm ischemia-reperfusion injury.
Forty-one consecutive primary kidney transplant patientsreceiving kidneys from deceased donors and experienc-ing DGF were enrolled in this observational study. Allpatients signed an informed consent form agreeing to
Materials and Methods
participate in the study. The presence of DGF was de-
Renal Ischemia-Reperfusion Injury Model
fined as the need for dialysis in the first week after trans-plantation. All patients included in the study were given
After approval by the ethical committee of the Ministry of
500 mg of methylprednisolone intraoperatively, followed
Health, ten 4-month-old female Large White pigs weight-
by 250 mg of prednisone daily, with the dose tapered to
ing 20 kg underwent experimental open surgical proce-
25 mg by day 8; 20 mg of a chimeric monoclonal anti-
dure under general anesthesia. The animals were fasted
CD25 antibody (Simulect, Novartis) intravenously on day
for 24 hours before induction of anesthesia. The electro-
0 and day 4; mycophenolate mofetil (Cell-Cept, Roche)
cardiogram, heart rate, hemoglobin saturation of oxygen,
1000 mg b.i.d and either cyclosporine A (Neoral, Novar-
respiratory gas composition, respiratory rate, tidal vol-
tis, C2 levels 800 to 1200 ng/ml) or tacrolimus (Prograf,
ume, airway pressure, systolic arterial blood pressure,
Astellas, through levels 8 to 12 ng/ml). According to our
and central venous pressure were continuously moni-
standard clinical protocol, a wedge biopsy before trans-
tored and automatically recorded (Ohmeda Modulus CD;
plantation was performed on all deceased donor kidneys.
Datex Ohmeda, Helsinki, Finland).
A second graft biopsy was performed 7 to 15 days (mean
The left renal artery and vein were isolated and a
10.2 days) after transplantation in all patients with DGF.
vessel loop was positioned around the renal artery with a
All patients with pretransplant panel reactive antibod-
right angle clamp. A renal biopsy was performed before
ies (PRA, Luminex, One Lambda, Montpellier France) ⬎
ischemia (T0). Then, the ischemic phase was induced
0%, or historical evidence of neoplasia, active systemic
(30 minutes) by pulling on the vessel loop. Five minutes
infection, or biopsy-proven acute rejection were ex-
before the end of the ischemia time, rhC1INH, isolated
cluded from the study. As control group, we used 10
from the milk of transgenic rabbits (Pharming), was di-
kidney graft recipients that underwent renal biopsy within
luted in saline solution and was injected in the ear vein of
15 days from transplantation for calcineuirin inhibitors
five animals (500 U/kg); in another group of five animals
toxicity. In DGF patients the graft biopsy revealed a dif-
an equal volume of vehicle was infused at the same time
fuse acute tubular necrosis with no evidence of acute
point (control group). Multiple biopsies were then per-
rejection including borderline changes and/or diffuse
AJP April 2010, Vol. 176, No. 4
peritubular capillary deposition of C4d. At the same time
glycerol (DakoCytomation, Carpintera, CA). Negative
no patient presented with an increase of PRA. The study
controls were obtained incubating serial sections with
was performed according to the principles of the Decla-
the blocking solution and then control irrelevant antibody.
ration of Helsinki and was approved by our EthicsCommittee.
Staining Scoring
Microscopy Study
The number of C4d⫹ peri-tubular capillaries and thenumber of CD163⫹, CD4a⫹, and CD8a⫹ infiltrating cells
Paraffin-embedded renal specimens from the animal
were evaluated in at least 10 high power (⫻630) fields
model and from renal biopsies were used for conven-
(hpf)/section by two independent observers blinded to
tional histological staining (H&E, periodic acid-Schiff).
the origin of the slides. The final counts were the mean of
Tubulointerstitial and glomerular lesions were evaluated
the two measures. In no case interobserver variability
using a qualitative analysis by two observers (M.R., G.G.)
was higher than 20%. Specific C4d immunostaining at
who were unaware of the origin of the slides.
the glomerular level was quantified using Adobe Photo-shop software and expressed as pixel/glomeruli.
Tissue Immunofluorescence and Confocal Laser
The primary antibodies used in this study recognized thefollowing antigens: CD8a (76-2-11, BD Biosciences, San
Jose, CA); CD4a (MIL17, AbD Serotec, Oxford, UK); CD163
Human and pig sections were double-stained for C1q/
(monocytes/macrophages, US biological, Swampscott,
C4d, MBL/C4d, and MASP2/MBL. The slides were incu-
MA); SWC3a (dendritic cells,16 74-22-15A, BD Bio-
bated with protein block serum-free (Dako) for 10 minutes
sciences); caspase-3 (Novus Biologicals, Inc, Littleton,
and 2% bovine serum albumin for 1 hour at room tem-
CO); C1q (R9/2, AbD Serotec); MBL (3E7, Hycult bio-
perature. Slides were then incubated for 1 hour at room
technology b.v., Uden, the Netherlands); C4d (Bio-
temperature with the specific antibodies. Slides were
medica Gruppe, Wien, Austria); Factor B (Novus Biologi-
then incubated with the appropriate secondary antibod-
cals); C9 neo antigen (aE11, Dako, Glostrup, Denmark);
ies (Alexa Flour 488 goat anti-rabbit and 555 anti-mouse,
MASP-2 (N-20, Santa Cruz Biotechnology, Santa Cruz, CA);
both from Molecular Probes, Eugene, OR). For Factor B,
and C3c (Dako Cytomation, Glostrup, Denmark). The cross
C5b-9, and SWC3a detection, slides were incubated with
reactivity was validated pre-incubating the specific antibod-
the specific antibody overnight at 4°C. After three washes
ies, before their use, with human peptides used to raise
in PBS, slides were incubated with the appropriate sec-
them. The pre-incubation completely abolished the specific
ondary antibody (Molecular Probes). All sections were
staining on swine tissue.
counterstained with TO-PRO-3 (Molecular Probes). Neg-ative controls were prepared with irrelevant antibody. The
sections were analyzed using the Leica TCS SP2 (Leica,Wetzlar, Germany) confocal laser-scanning microscope.
C4d, Caspase3
The number of SWC3a⫹ cells was measured in at least10 high power (⫻630) fields/section by two independent
Two m-thick sections of human and pig paraffin-
observers blinded to the origin of the slides. The final
embedded renal tissue were rehydrated by a series of
counts were the mean of the two measures. In no case
xylene and graded alcohol washes. After antigen re-
interobserver was variability higher than 20%.
trieval, the sections were blocked with bovine serumalbumin 2% for 1 hour at room temperature. The sectionswere incubated with the primary antibodies (anti-C4d or
anti-caspase3) and detected by the Dako EnVision G/2System (Dako). The sections were counterstained with
Data are expressed as the mean ⫾ SD. Difference be-
Mayer's hematoxylin and mounted with glycerol. Nega-
tween groups was analyzed by paired
t-test analysis. A
tive controls were obtained incubating serial sections
P value ⬍0.05 was considered statistically significant.
with the blocking solution and irrelevant antibody.
CD163, CD4a, CD8a
Cryostat sections (4 m-thick) of frozen pig kidneys
Activation of Classical, Lectin, and Alternative
were stained for infiltrating cells markers. Slides were
Pathways in a Swine Model of
prepared and incubated with specific antibodies. Endog-
enous peroxidase activity was blocked by 3% H O for 7
minutes. The tissue samples were then incubated with
We first investigated the mechanism of complement cas-
Dako Real EnVision Detection System (Dako). The re-
cade activation in the swine model of warm ischemia-
action was visualized by a brown precipitate, counter-
reperfusion⫺induced renal injury. The terminal comple-
stained with Mayers hematoxylin (blue) and mounted with
ment complex, C5b-9, was barely detectable in the basal
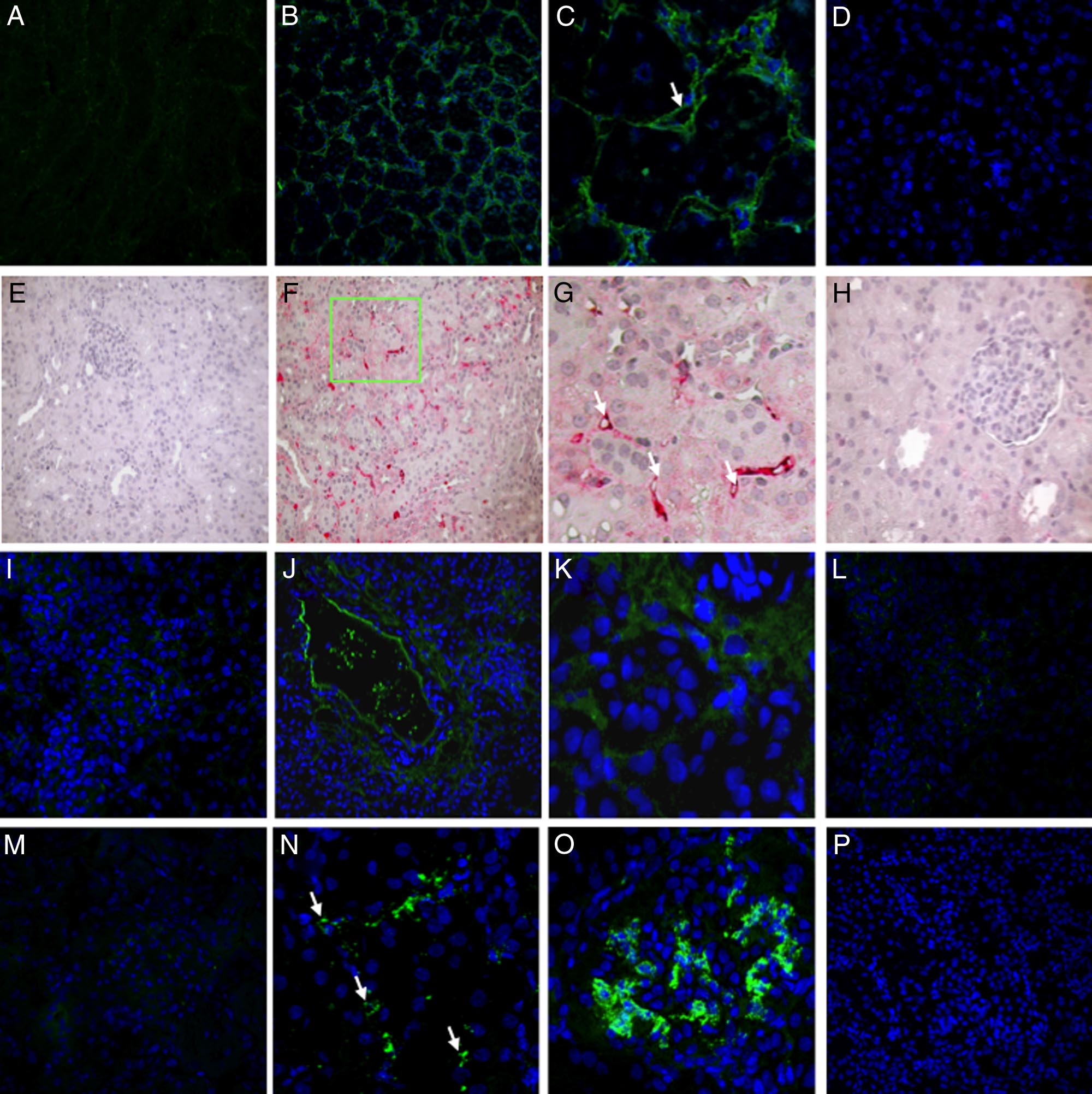
C1-Inhibitor in Renal I/R Injury
AJP April 2010, Vol. 176, No. 4
Figure 1. Classical, alternative, and lectin pathway activation in a pig model of ischemia-reperfusion injury. Frozen pig kidney sections were examined for
expression of the membrane attack complex (C5b-9) by immunofluorescence and confocal analysis (A⫺D). C5b-9-deposition, completely absent before ischemia
(A), was significantly up-regulated 30 minutes after reperfusion (B), in particular at the peritubular and capillary level (C, arrow) and disappeared 24 hours after
reperfusion (D). C4d deposition was evaluated by immunohistochemistry on paraffin-embedded kidney sections (E⫺H). Specific C4d staining was not observed
at T0 (E). Thirty min of reperfusion induced a diffuse deposition of C4d (F) with a specific localization at the peritubular capillaries (G, arrows) that disappeared
at 24 hours (H). Factor B deposition was investigated by immunofluorescence and confocal analysis (I⫺L). Factor B deposition, completely absent before ischemia
(I), was significantly up-regulated 30 minutes after reperfusion (J), particularly at the interstitial and tubular level (K), and disappeared 24 hours after reperfusion
(L). Complement C3 deposition was investigated by immunofluorescence and confocal analysis (M⫺P). Complement C3 deposition, absent before ischemia (M),
was significantly increased 30 minutes after reperfusion (N⫺O; N, peritubular capillaries indicated by arrows), and disappeared 24 hours after reperfusion (P).
In confocal microscopy images nuclei were stained with TO-PRO-3 (blue).
biopsies (Figure 1A). Thirty minutes of ischemia caused a
Subsequently, we investigated the presence of C4d in
diffuse deposition of C5b-9 at 30⬘ of reperfusion (Figure
kidney biopsies before and after reperfusion. As shown in
1, B and C), which disappeared after 24 hours (Figure
Figure 1E, before the induction of ischemia, C4d deposits
1D). The complement terminal complex was localized at
were undetectable in renal biopsies, whereas after 30
the peritubular level (Figure 1B) as well as within the
minutes of reperfusion, we observed a significant depo-
peritubular capillaries along the endothelial cell layer
sition of C4d both at tubulointerstitial (Figure 1, F and G)
(Figure 1C, arrow).
and glomerular (data not shown) levels. In both cases
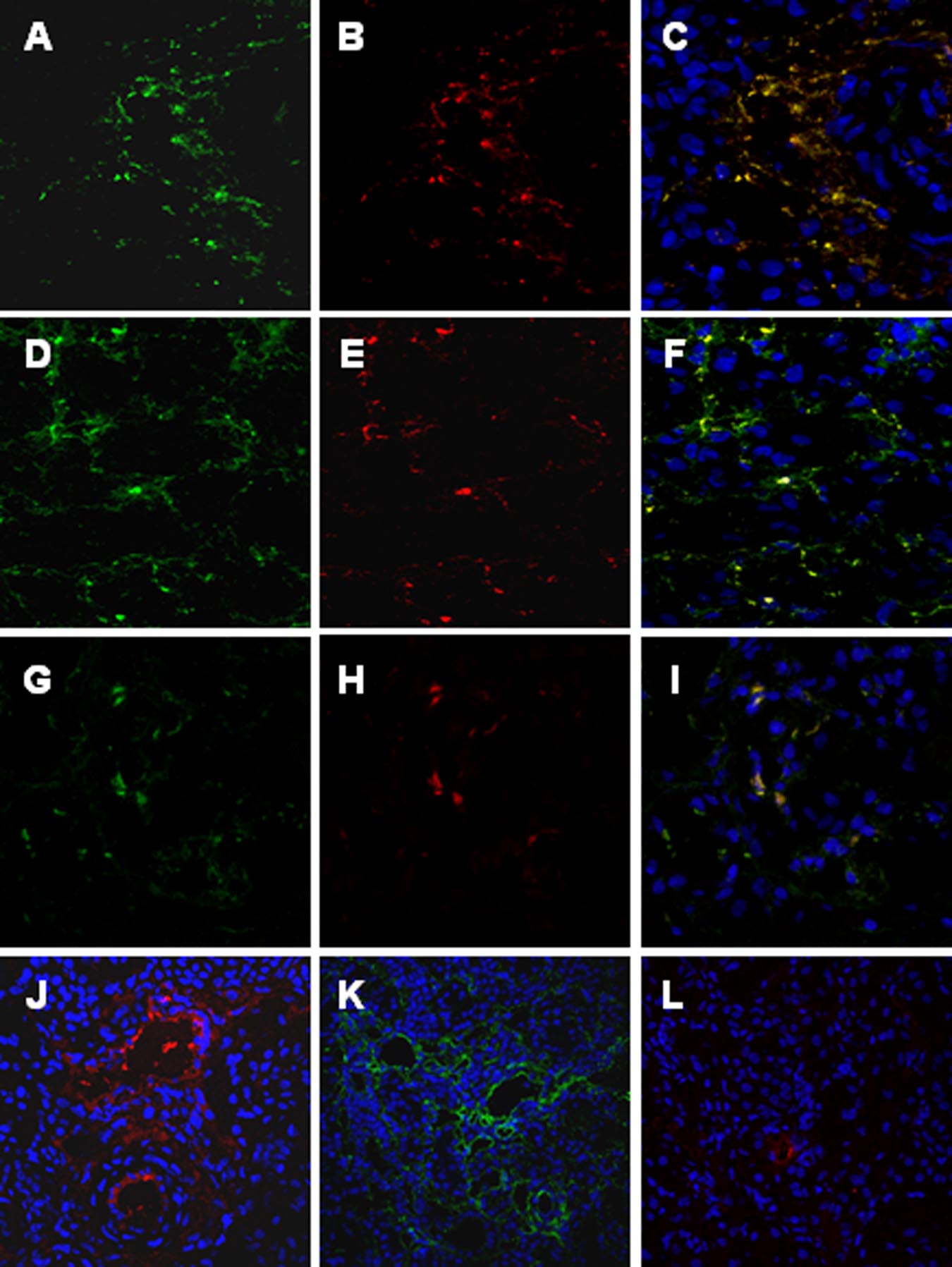
AJP April 2010, Vol. 176, No. 4
Figure 2. C4d deposition in the swine model of
ischemia-reperfusion-induced renal injury is due
to both classical and lectin pathways activation.
Co-localization of C4d with C1q (A⫺C), MBL
(D⫺F), and MBL/MASP-2 (G⫺I) was investi-
gated on frozen kidney sections by immunoflu-
orescence/confocal microscopy. A: Interstitial
and capillary deposition of C4d (green), (B) in-
terstitial deposition of C1q (red); (C) co-deposi-
tion of C1q-C4d at interstitial level (merge).
D: Interstitial and capillary deposition of C4d
(green), (E) interstitial and capillary deposition
of MBL (red); (F) co-deposition of MBL-C4d at
interstitial and capillary level (merge). The same
pattern of co-deposition was also observed for
MASP-2/MBL (G⫺I). The complement cascade
was predominantly activated by the lectin and
alternative pathways at 15 minutes of reperfu-
sion. MBL, factor B and C1q deposits were in-
vestigated by immunofluorescence/confocal mi-
croscopy. The extent of MBL (J) and factor B (K)
deposition was significantly higher than the one
observed for C1q (L). Nuclei were stained with
TO-PRO-3 (blue).
C4d deposits were localized at the endothelial cells sur-
C4d deposits in our swine model of ischemia/reperfusion
face (Figure 1G, arrows). As described for C5b-9, the
colocalized with either C1q or MBL. We observed the
deposition of C4d was markedly reduced at 24 hour
co-deposition of C4d with both C1q (Figure 2, A⫺C) and
(Figure 1H). Moreover, we confirmed the activation of the
MBL (Figure 2, D⫺F). To further confirm the activation of
alternative pathway in this setting. Indeed, factor B dep-
the lectin pathway we studied also the deposition of
osition, completely absent in the kidney before ischemia
MASP-2. As shown in Figure 2, G⫺I, MASP-2 colocalized
(Figure 1I), was significantly increased after 30 minutes of
reperfusion (Figure 1, J and K). Factor B was mainly
To clarify which of the three complement pathways
present at the interstitial and tubular level (Figure 1J), as
was primarily activated in our experimental model, we
previously described. The alternative pathway was also
investigated C1q, MBL, and factor B deposition 15 min-
transiently activated in this model of renal ischemia-
utes after reperfusion. The extent and diffusion of MBL
reperfusion injury, since factor B deposits were virtually
(Figure 2J) and factor B (Figure 2K) deposits were sig-
absent after 24 hours of reperfusion (Figure 1L). Finally,
nificantly more pronounced then C1q deposition (Figure
the C3c deposits appeared with the same time and spa-
2L), suggesting a predominant role of the lectin and
tial pattern of the previously described complement fac-
alternative pathways in this setting.
tors (Figure 1, M⫺P). C3c was localized on peritubularcapillaries (Figure 1N, arrows), and on glomerular endo-
rhC1INH Treatment and C4d Deposition
thelial cells (Figure 1O).
Since C4 deposition could be due to either classical or
On the basis of our data in the swine model of ischemia-
lectin pathway activation, we then investigated whether
reperfusion⫺induced renal injury, we hypothesized that
C1-Inhibitor in Renal I/R Injury
AJP April 2010, Vol. 176, No. 4
rhC1INH Treatment and Infiltrating InflammatoryCells
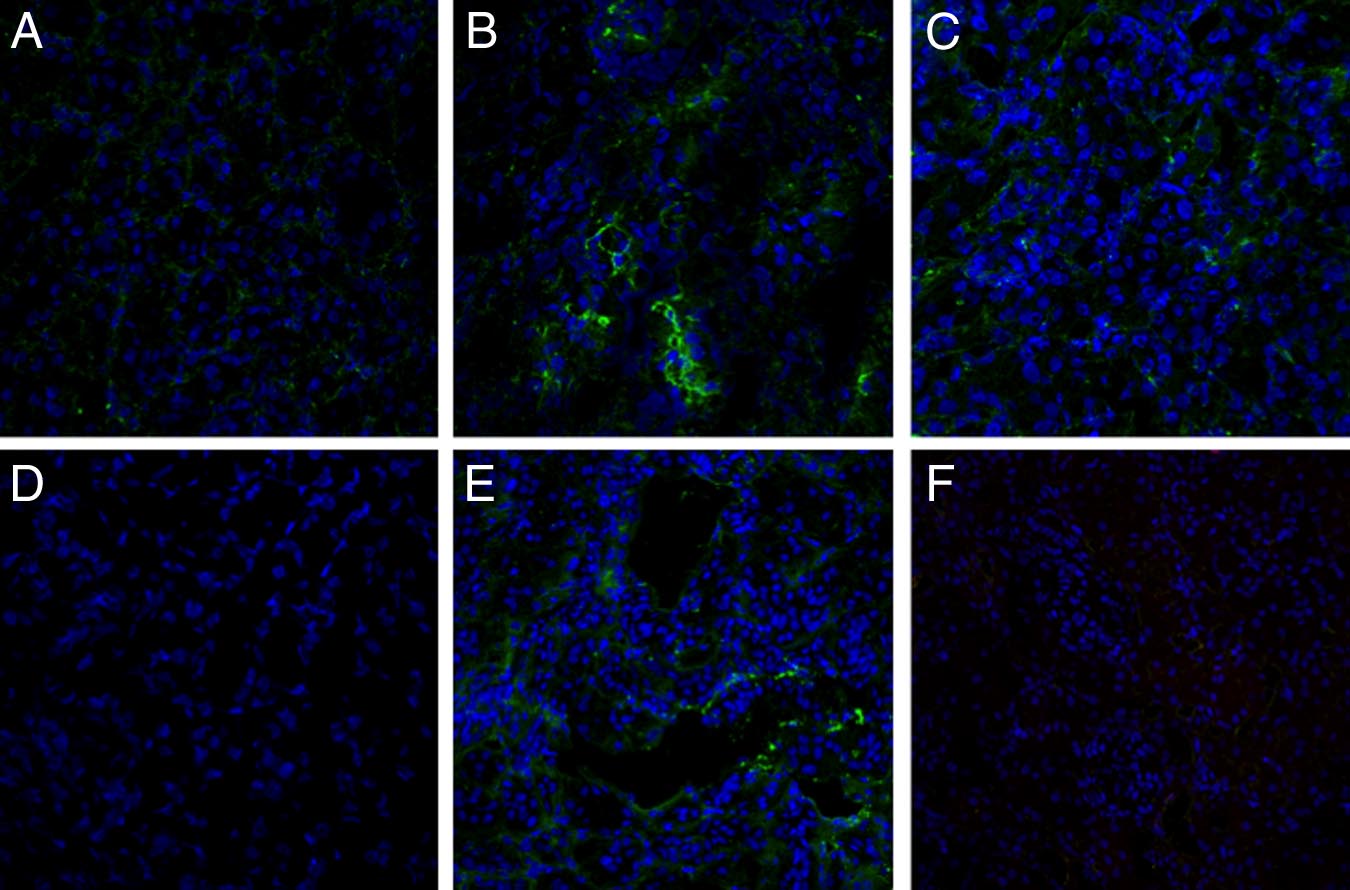
Next, we investigated the ability of rhC1INH to reduce therecruitment of infiltrating inflammatory cells, one of thehallmarks of ischemia-reperfusion injury, by inhibitingcomplement activation. Control pigs showed significantrecruitment of infiltrating CD163⫹ monocyte/macro-phages (Figure 6, A and B), whereas rhC1INH-treatedpigs showed a significant decrease of CD163⫹ cellsthroughout the observation period (Figure 6, C and D). Inaddition, we investigated the presence of dendritic cellsusing a specific polyclonal antibody against SWC3a⫹.
As previously reported,16 infiltrating dendritic cells weresignificantly increased after reperfusion in the vehicle-treated animals (Figure 6, E and F). Interestingly,rhC1INH-treated pigs showed a significant reduction ininfiltrating SWC3a⫹ cells throughout the observationperiod (Figure 6, G and H).
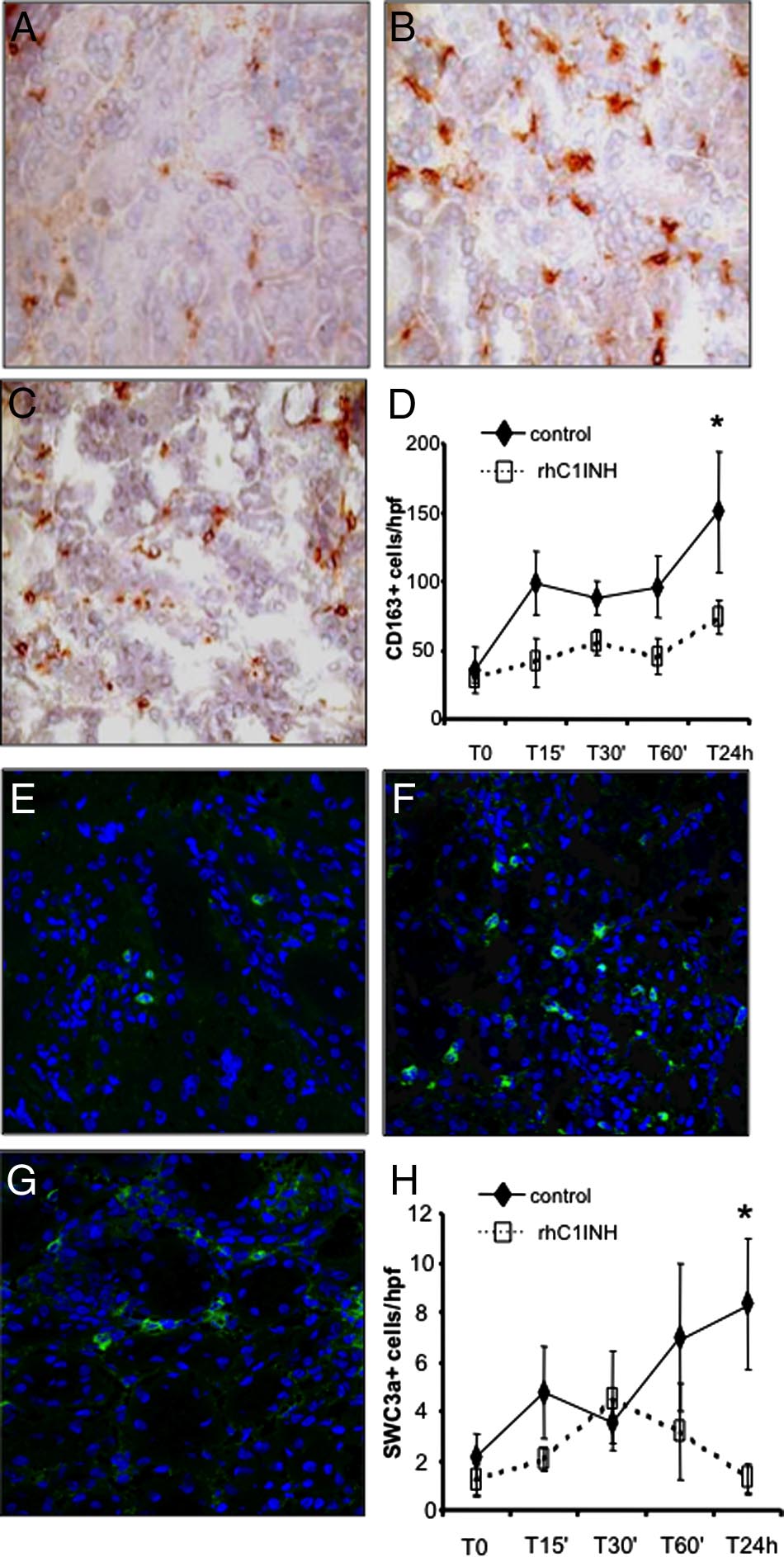
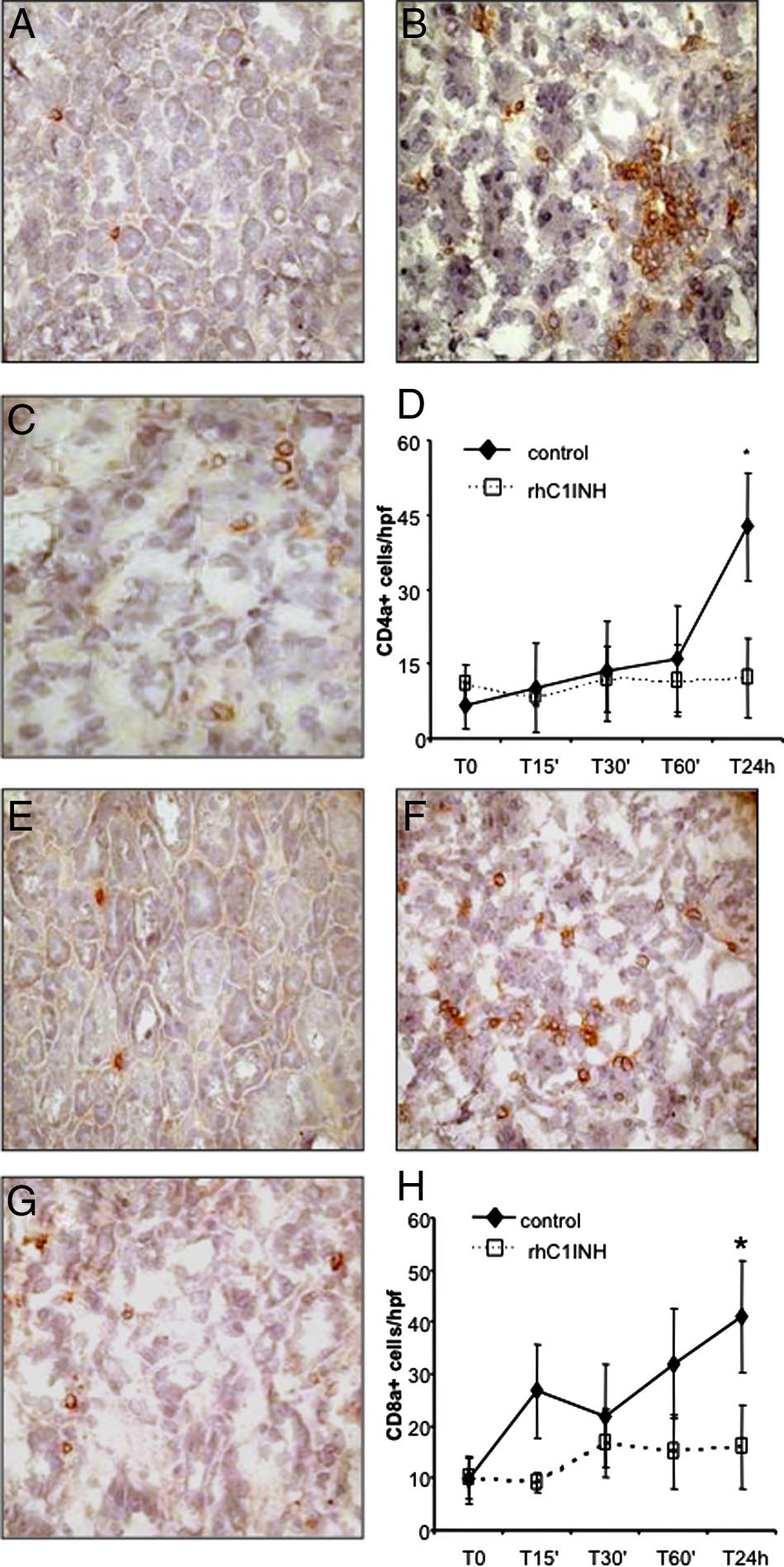
Finally, we investigated whether the recruitment of T
lymphocytes might also be influenced by rhC1INHtherapy. As shown in Figure 7A, rare CD4⫹ T cells werepresent within the tubulointerstitial area before the in-duction of ischemia-reperfusion. After 24 hours of
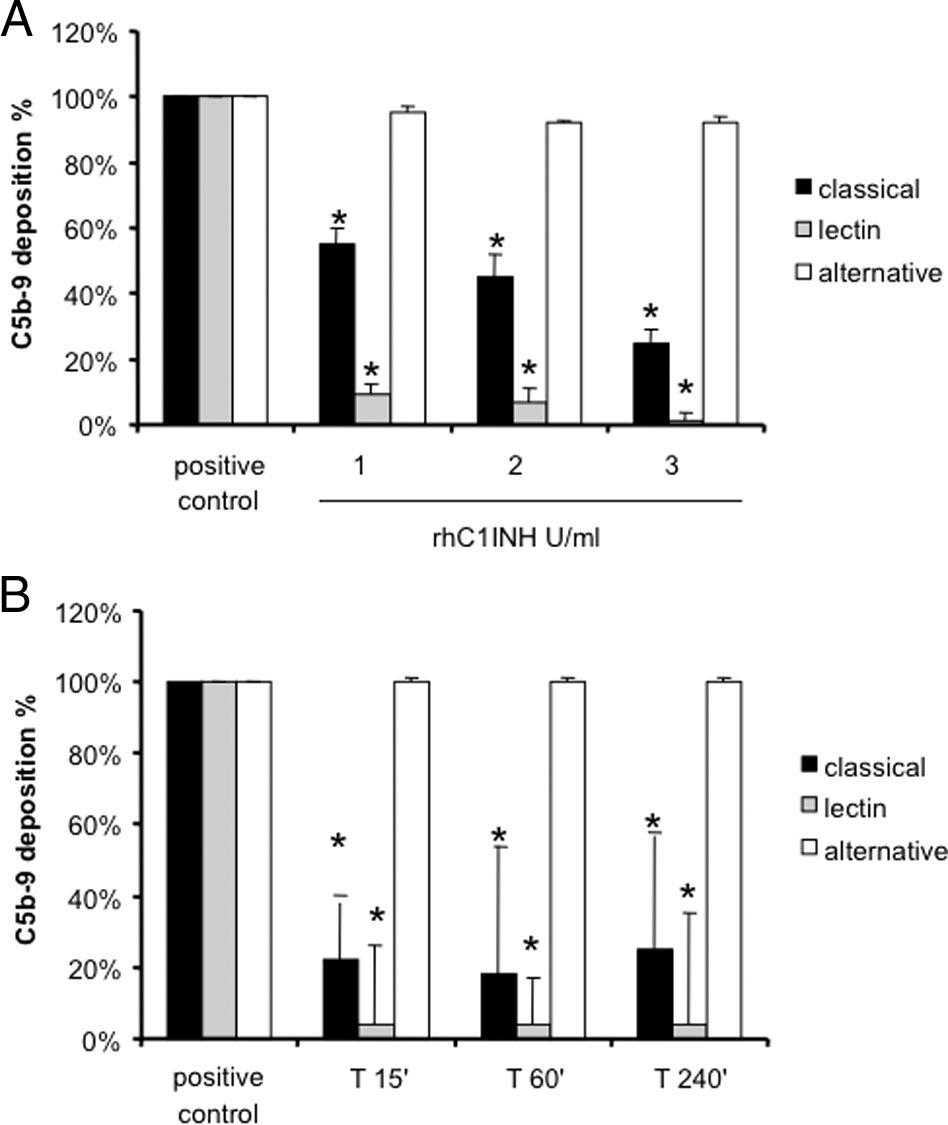
Figure 3. Assessment of functional activity of classical, alternative, and lectin
reperfusion (Figure 7B), there was a major influx of
pathways in pig sera. A: Increasing concentrations of rhC1INH were incu-
these cells. The recruitment of CD4⫹ T cells during the
bated with sera from healthy pigs to evaluate the capacity to inhibit theclassical and lectin pathways (Wielisa, Wieslab). *P ⬍ 0.05 versus positive
24 hours was greatly reduced in rhC1INHtreated ani-
control (pig sera with vehicle). B: Three healthy pigs received 500 U/kg of
mals (Figure 7, C and D).
rhC1INH; then, serum samples were collected at different time points andevaluated for complement activation by Wielisa. *P ⬍ 0.05 versus positive
CD8⫹ T lymphocytes were also absent in the normal
kidney (Figure 7E) and we observed a significant tubu-lointerstitial influx of these cells, most notable 24 hoursafter reperfusion, in the vehicle-treated animals (Figure
rhC1INH, a specific inhibitor of both classical and lectin
7F). In rhC1INH-treated animals this increase was not
pathways, might reduce the activation of the complement
observed (Figure 7, G and H).
cascade in this setting. We first tested in vitro the ability ofrhC1INH to inhibit the classical and lectin pathways in pigsera. Increasing concentrations of rhC1INH were able toreduce more than 50% of C5b-9 generation due to the
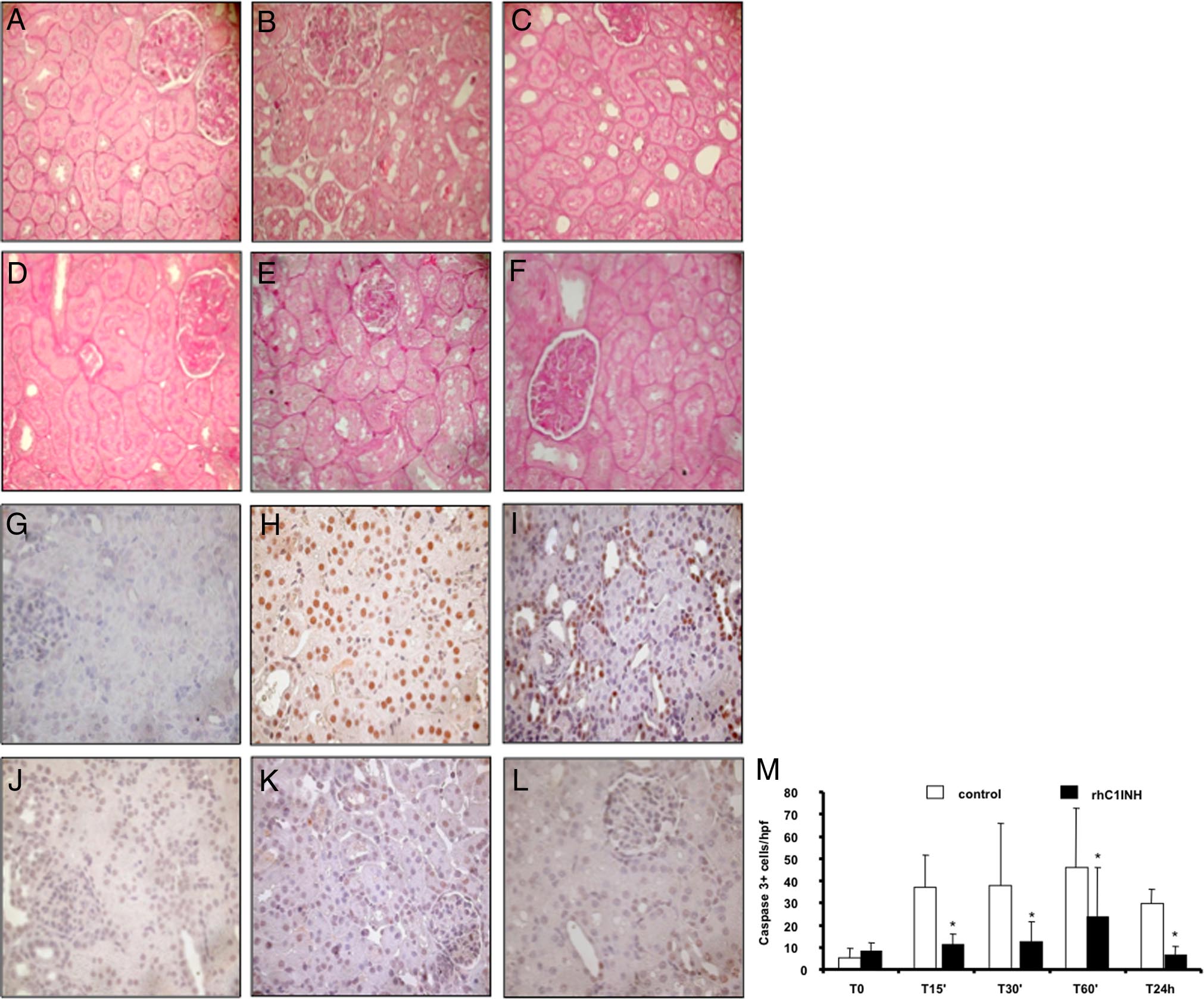
rhC1INH Treatment and Tubular Damage
activation of the classical pathway in vitro (Figure 3A).
Interestingly, inhibition of the lectin pathway reached
Together with the recruitment of infiltrating inflamma-
100% at the concentration of 3 U/ml (Figure 3A). In ad-
tory cells, the other main feature of ischemia-reperfu-
dition, we tested ex vivo the ability of the rhC1INH to
sion injury is tubular epithelial cell damage. On routine
modulate the lectin and classical pathway of complement
histological evaluation, untreated animals developed
activation. We infused 500 U/kg i.v. in three animals and
tubular vacuolization, loss of brush border, and lumen
collected serum samples at different time points. The
dilatation after 60 minutes of reperfusion that increased
drug was able to significantly reduce both lectin and
in severity up to 24 hours postreperfusion (Figure 8,
classical pathway activation up to 240 minutes after infu-
A⫺C). These changes were markedly reduced in
sion (Figure 3B).
rhC1INH-treated animals (Figure 8, D⫺F). We then ex-
We then moved to the experimental model and ob-
amined tubular cell apoptosis using caspase-3 expres-
served that the animals receiving rhC1INH i.v. 5 minutes
sion as a specific marker. Caspase-3⫹ cells, rarely
before reperfusion presented a clear reduction in C4d
present within tubular sections at baseline (Figure 8G),
deposition at the tubulointerstitial and glomerular (Figure4, A⫺C) levels compared with control animals (Figure 4,
were dramatically increased 60 minutes and 24 hours
D⫺F). Quantification of C4d deposit at tubulointerstitial
after reperfusion (Figure 8, H and I, respectively).
and glomerular levels demonstrated a significant differ-
Interestingly, this phenomenon was completely abol-
ence between treated and untreated animals at multiple
ished in rhC1INH-treated animals (Figure 8, J, K,
postreperfusion time points (Figure 4, G and H, respec-
and L). Quantification of caspase-3⫹ cells demon-
tively). In addition, we also observed a marked reduction
strated that the difference between the two groups
in the deposition of C5b-9 (Figure 5, A⫺C) and MASP2
was significant at all of the time points evaluated (Fig-
(Figure 5, D⫺F) in rhC1INH-treated animals.
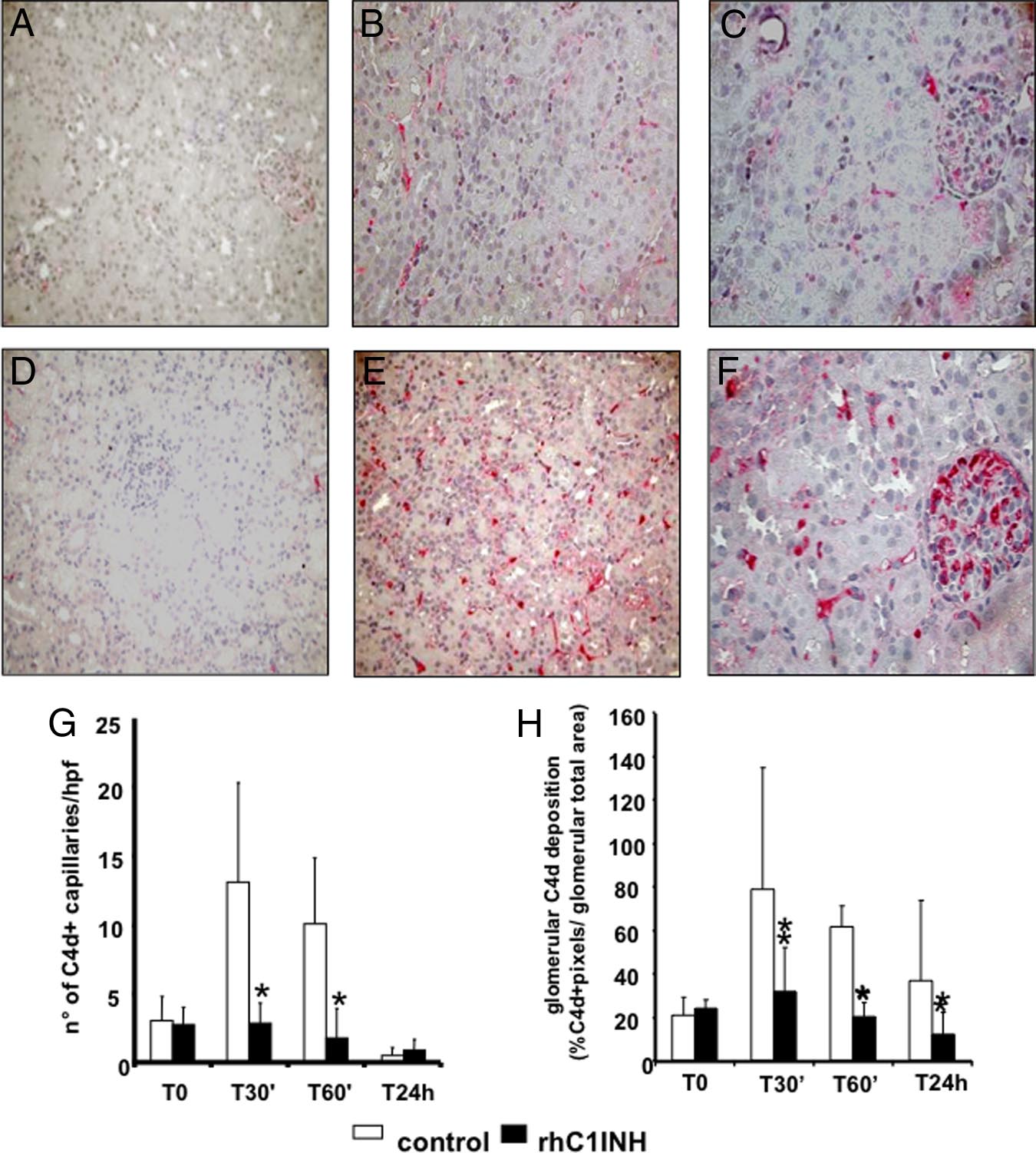
AJP April 2010, Vol. 176, No. 4
Figure 4. Inhibition of C4d deposition by
rhC1INH treatment in a swine model of isch-
emia-reperfusion-induced renal injury. C4d de-
posits were evaluated by immunohistochemistry
on paraffin-embedded kidney sections from an-
imals treated either with rhC1INH (500 UI/kg,
A⫺C) or vehicle (D⫺F). The C4d deposits, ab-
sent before ischemia induction (A, D), remained
significantly lower in the treated group both at
the tubulointerstitial (B) and glomerular (C)
level compared with untreated animals (tubulo-
interstitial: E; glomerular: F) Original magnifica-
tion ⫻400x. G: Quantification of peritubular
capillary C4d⫹ at the different times of reperfu-
sion in treated (n ⫽ 5) and control animals (n ⫽
5). Results are expressed as the mean ⫾ SD of
C4d⫹ capillary/high power field (hpf). *P ⬍ 0.05
versus time-matched vehicle-treated animals.
H: Quantification of glomeruli C4d⫹. Results
are expressed as the mean ⫾ SD of glomeruli
C4d⫹. *P ⬍ 0.05 vs. time-matched vehicle-treated
animals.
Analysis of Complement Activation in Patients
tion of the complement system. Forty percentage of DGF
patients showed focal C4d deposits in peritubular capil-lary, 34% showed deposition at the tubulointerstitial level,
Biopsy specimens from 41 transplant recipients with DGF
and 41% showed deposition at the glomerular capillary
were analyzed to detect fragments derived from activa-
level. Interestingly, none of the patients with C4d deposits
Figure 5. Inhibition of C5b-9 (A⫺C) and MASP-2
(D⫺F) deposition by rhC1INH treatment in a
swine model of ischemia-reperfusion⫺induced re-
nal injury. C5b-9 and MASP-2 deposits were inves-
tigated by confocal microscopy. Both were absent
before ischemia (A and D, respectively) and were
up-regulated 30 minutes after reperfusion (B and
E, respectively) in vehicle-treated animals. C1INH
infusion caused a marked reduction in the depo-
sition of both, C5b-9 (C) and MASP-2 (F).
C1-Inhibitor in Renal I/R Injury
AJP April 2010, Vol. 176, No. 4
Figure 7. Modulation of lymphocytes infiltration by rhC1INH treatment in a
swine model of ischemia-reperfusion⫺induced renal injury. CD4a expression
Figure 6. Modulation of monocyte and dendritic cell infiltration by
was evaluated in kidney tissue cryosection by immunohistochemistry. Very few
rhC1INH treatment in a swine model of ischemia-reperfusion-induced
infiltrating CD4a⫹ cells were observed before ischemia within the interstitial
renal injury. CD163 (a monocyte/macrophage marker) expression was
compartment (A). Twenty-four hours of reperfusion induced a significant re-
evaluated in kidney tissue cryosection by immunohistochemistry. A low
cruitment of CD4a⫹ cells (B) in vehicle-treated animals, whereas rhC1INH
number of infiltrating CD163⫹ cells was observed before ischemia within
administration caused a marked reduction in the number of infiltrating CD4a⫹
the interstitial compartment (A). Twenty-four hours of reperfusion in-
lymphocytes (C). D: Quantification of CD4a⫹ cells at different time points after
duced a significant recruitment of CD163⫹ cells (B) in vehicle-treated
induction of ischemia-reperfusion. Results are expressed as the mean⫾s.d of
animals, whereas rhC1INH administration caused a marked reduction in
CD4a⫹cells/high power field (hpf). *P ⬍ 0.05 versus T0 and versus T24 hours of
the number of infiltrating monocytes (C). D: Quantification of CD163⫹
rhC1INH-treated animals. CD8a expression was investigated in kidney tissue
cells at different time points after induction of ischemia-reperfusion.
cryosection by immunohistochemistry. Very few infiltrating CD8a⫹ cells were
Results are expressed as mean ⫾ s.d of CD163⫹cells/high power field
observed before ischemia within the interstitial compartment (E). Twenty-four
(hpf). *P ⬍ 0.05 versus T0 and versus T24 hours of rhC1INH-treated
hours of reperfusion induced the recruitment of CD8a⫹ cells (F) in vehicle-
animals. Original magnification ⫻400. The protein expression of SWC3 (a
treated animals, whereas rhC1INH administration caused a marked reduction in
marker of dendritic cells) was investigated by immunofluorescence/con-
the number of infiltrating CD8a⫹ T cells (G). H: Quantification of CD8a⫹ cells
focal microscopy. Very few infiltrating SWC3A⫹ cells were observed
at different time points after induction of ischemia-reperfusion. Nuclei were
before ischemia within the interstitial compartment (E). Twenty-four
counterstained by hematoxylin. Original magnification ⫻400. *P ⬍ .05 versus T0
hours of reperfusion induced a significant recruitment of SWC3A⫹ cells
and versus T24 hours of rhC1INH-treated animals.
(F) in vehicle-treated animals, whereas rhC1INH infusion caused a
marked reduction in the number of infiltrating dendritic cells (G). TO-
PRO-3 (blue) was used to counterstain nuclei. Original magnification
showed histological signs of acute rejection. As shown
⫻630. H: Quantification of SWC3a⫹ cells during ischemia-reperfusion.
in Figure 9A, C4d deposits were present diffusely at
Results are expressed as mean ⫾ s.d of SWC3A⫹ cells/high power field
the tubulointerstitial level and focally within the peritu-
(hpf). *P ⬍ 0.05 versus T0 and versus T24 hours of rhC1INH-treatedanimals.
bular capillaries (Figure 9B). Moreover, the deposits
AJP April 2010, Vol. 176, No. 4
Figure 8. Modulation of tubulointerstitial damage and
tubular cell apoptosis by rhC1INH treatment in a swine
model of ischemia-reperfusion⫺induced renal injury.
Compared with basal conditions (A) ischemia-reperfu-
sion induced an evident vacuolization and an early cel-
lular infiltrate associated with interstitial edema at the
tubulointerstitial level (B: 60 minutes and C: 24 hours of
reperfusion). Significantly less edema and tubular dam-
age were present in pigs treated with rhC1INH (D: base-
line, E: 60 minutes, and F: 24 hours of reperfusion).
Nuclear protein expression of caspase-3 (a specific
marker of late cell apoptosis) was evaluated by immu-
nohistochemistry on paraffin-embedded tissue sections.
Very few caspase-3⫹ cells were observed before isch-
emia (G). An increase in the number of apoptotic tubular
cells was observed after 60 minutes (H) and 24 hours (I)
of reperfusion in vehicle-treated animals. RhC1INH infu-
sion caused a dramatic reduction in caspase-3⫹ cells at
both time points (J: baseline, K: 60 minutes, and L: 24
hour). M: Quantification of caspase-3⫹ cells after re-
nal ischemia-reperfusion injury. Results are expressed
as mean ⫾ SD of caspase-3⫹ cells/high power fields
(hpf). *P ⬍ 0.05 versus vehicle-treated animals (mag-
nification ⫻400).
were also present in the glomerular capillaries (Figure
stitial damage. Finally, we demonstrated that these two
9C). On the contrary, control patients showed no C4d
pathways were also activated in renal transplant recipi-
deposits (Figure 9D).
ents suffering from DGF.
To determine whether C4d deposits were due to acti-
DGF is the primary early complication seen in kidney
vation of classical or lectin pathways, we performed dou-
transplant recipients, affecting more than 25% of pa-
ble staining for C1q/C4d and MBL/C4d. Patients with
tients.1 This condition is associated with an increased
DGF had co-deposition of both C1q and C4d (Figure 9, E,
incidence of acute rejection episodes and worse long-
G, and I), indicating that activation of complement occurred
term graft survival. In addition, the increasing use of
via the classical pathway. We also observed a clear colo-
expanded criteria donors inevitably results in a significant
calization of MBL and C4d (Figure 9, F, H, and J), suggest-
increase in the incidence of DGF, with the foreseeable
ing the concurrent activation of the lectin pathway.
consequences for the National Health Systems of in-creasing costs for kidney transplantation programs. Todate we do not have specific therapeutic strategy to
prevent DGF or to reduce its length.3,17 Thus, this early
In the present study we demonstrated for the first time
post-transplant condition remains a significant concern
that activation of the complement system occurs with the
for transplant physicians. An increasing amount of evi-
involvement of both classical and lectin pathways in a
dence derived from animal models suggests that the
swine model of renal ischemia-reperfusion injury. We
injury secondary to the ischemia-reperfusion process
demonstrated that therapeutic inhibition of classical and
might play a pivotal role in the pathogenesis of this form
lectin pathways by rhC1INH produced a significant re-
of acute renal failure.1
duction in complement deposition, with decreased re-
One of the most prominent events occurring in ische-
cruitment of infiltrating inflammatory cells and tubulointer-
mia-reperfusion is activation of the complement cas-
C1-Inhibitor in Renal I/R Injury
AJP April 2010, Vol. 176, No. 4
cade.4,5,18,19 The involvement of complement has beenshown in several animal models, where deficiency incomplement components significantly reduced the dam-age associated with ischemia-reperfusion (reviewed bySacks et al).4 In agreement with these observations, theuse of complement inhibitors was effective in limitingischemia-reperfusion damage in normal animals.9,10These data strongly suggest that the inhibition of com-plement may represent a potential target for preventingtissue damage.6,11
Interestingly, data available so far indicate that, in con-
trast to ischemia-reperfusion in the intestine, heart, andlung, complement activation due to ischemia-reperfusionin kidneys occurs almost exclusively through the alterna-tive pathway.4,5,14,19 C4 and Ig deficient mice were notprotected from ischemia-reperfusion injury.8,20 On thecontrary, factor B deficient mice were resistant to ische-mia-reperfusion⫺induced renal damage.21 Taken as awhole, these observations indicate that neither the clas-sical nor the lectin pathway of complement is involved inrenal injury due to ischemia-reperfusion. However, all ofthe studies present in the literature were conducted inrodents and, so far, no data are available from largeranimals.
In our swine model of renal ischemia-reperfusion in-
jury, we found C4d deposits primarily present at theperitubular and glomerular capillary levels, indicating thatthe endothelial cells might play a pivotal role in the acti-vation of complement. Ischemia can induce a lower re-sistance to complement activation in endothelial cells,leading to presentation of complement-activating sur-faces and autoantigens.22 MBL can bind to exposedcytokeratin on hypoxic endothelial cells, causing localactivation of the complement cascade through the lectinpathway. Indeed, it has been shown that patients withhigh circulating concentrations of MBL have a poor out-come after renal transplantation.23 Moreover, C4d is ac-tually seen as a specific marker of antibody-mediatedclassical pathway activation,24 being stable at the renallevel for a long period of time because of covalentlylinkages. On the contrary, in our model we showed thatC4d deposition was present in the early period afterischemia-reperfusion damage and disappeared at 24hours postreperfusion and, in this setting, was mainly dueto the activation of the lectin pathway. This led us tohypothesize that complement activation after ischemia-reperfusion on ischemic or necrotic cells is not sustainedin the long term, since these cells can be efficientlycleared by mononuclear phagocytes by 24 hours. This isconfirmed by the fact that, in the swine model, we ob-served the disappearance of C5b-9 and a significantreduction of C4d deposition at 24 hours. This transientdeposition of C4d in the animal model might also explain
Figure 9. C4d deposition in graft biopsies of patients with DGF. C4d deposits
the focal deposition of this complement component ob-
were evaluated by immunohistochemistry on paraffin-embedded graft tissue
served 7 to 10 days after reperfusion in transplant recip-
as described in the methods section. C4d deposits were observed in patients
with DGF (A, B, and C) at interstitial (A), peri-tubular capillary (B), and
ients as well as the observation that C4d could not be
glomerular capillary (C) level, whereas no C4d was found in control grafts
detected in all DGF patients.
(D). Co-localization of C4d with C1q (E–G–I) and MBL (F–H–J) was inves-
tigated by immunofluorescence/confocal microscopy. C4d (green, E) co-
On the basis of the data on C4d deposition in our
localized with C1q (red, G) at peri-tubular capillary sites (merge, I). C4d
model, we investigated whether rhC1INH, a drug cur-
deposits (green, F) co-localized with MBL (red, H) at the same site (merge,
rently used in preclinical trials in patients suffering from
J). TO-PRO-3 (blue) was used to stain nuclei. Original magnifications were
either (A–D) ⫻400 or (E–J) ⫻630.
hereditary angioedema due to genetic deficiency of
AJP April 2010, Vol. 176, No. 4
C1INH,25,26 was able to interfere with complement acti-
complement in the specific population of DGF patients.
vation in renal ischemia-reperfusion injury. Previous re-
Our data seems to be conflicting with a previous report,
search has shown that C1INH reduces complement ac-
which, however, analyzed complement activation in
tivation with significant protection of the myocardium and
clearly different population of transplanted patients.41– 43
brain from ischemic damage.27–30 Moreover, C1INH lim-
In particular, Haas et al41 reported no C4d deposits in
ited leukocyte adhesion and neutrophil infiltration in a
postimplantation biopsies performed 1 hour after reper-
model of ischemia-reperfusion in the liver.31–33 We ob-
fusion. Although most of their cases were transplanta-
served that rhC1INH was very effective in limiting com-
tions from living donors, with a very limited cold ischemia
plement activation in renal tissue, as showed by the
time, they also reported cases of deceased donor kidney
dramatic reduction of C4d and C5b-9 deposition. It is well
transplantations with over 30 hours of cold ischemia. The
known that early activation of complement leads to the
human model used by these authors closely resembles
release of active substances including C3a and C5a that
our experimental model, however also in our study we
can increase the recruitment of inflammatory cells.6,10,34
observed few differences between the complement acti-
As expected, pigs treated with rhC1INH showed reduced
vation observed in DGF patients and in the swine model.
infiltrating CD163⫹ monocyte/macrophages and SWC3a⫹
Indeed, in one third of the human subjects included in our
dendritic cells. Importantly, we observed that infiltrating T
study C4d staining was observed at 7 to 15 days after
lymphocytes were also significantly reduced in treated pigs.
transplantation, whereas in the swine model most of the
We hypothesize that this effect might be the result of a
specific deposits were gone by 24 hours. This observa-
double action of rhC1INH. Indeed, this serine protease in-
tion would suggest a different kinetics of complement
hibitor, next to the inhibition of complement activation, it has
activation in human subjects. Thus, it is conceivable that
been shown to interfere with selectin/integrin interaction,
the one hour postimplantation biopsy performed by Haas
therefore reducing the recruitment of leukocytes at the site
et al may have missed the complement priming induced
of inflammation.30
by ischemia-reperfusion in this setting. These apparently
In addition, it is well know that complement activa-
conflicting results from different studies with limited pa-
tion can induce resident cell apoptosis and necrosis by
tients' populations suggest the need for a large observa-
caspase activation through mediators such as C5b-9.35–38
tional study to confirm the value and the specificity of C4d
Indeed, we found the appearance of caspase-3⫹ tubular
deposition in peritransplant graft biopsies.
epithelial cells in the tubulointerstitial region in untreated
In conclusion, our data suggest a key role for thera-
pigs. The inhibition of complement by rhC1INH in this set-
peutic inhibition of classical and lectin pathway of com-
ting was sufficient to limit the induction of apoptosis to very
plement activation in the prevention of ischemia-reperfu-
low level, thus preventing ischemia-reperfusion⫺induced
sion injury in the transplanted kidney. On the basis of our
tubular damage. The reduced presence of infiltrating T cells
observation, the use of rhC1INH deserves further clinical
and dendritic cells associated with a marked decrease of
studies in transplant recipients at high risk for DGF, since
tubular cell damage might have a beneficial immunological
it may reduce the incidence and/or improve the clinical
effect in the setting of renal transplantation, limiting the
course of this common early post-transplant complica-
immunogenicity of the injured kidney, and reducing both
tion, potentially influencing long-term graft function and
direct and indirect alloantigen presentation. Despite the
significant protective effects demonstrated by this drug inpreserving the histology of ischemic kidney in our model,we believe that further clinical investigations are warranted
to evaluate the effects of rhC1INH infusion on renal functionin ischemia-reperfusion⫺induced renal injury.
We thank Claudia Curci, Nicoletta Fiore, Michelangelo
Finally, in the present study we identified clear signs of
Corcelli, and Vincenzo Gesualdo, from the Renal, Dialy-
involvement of the classical and lectin pathways in pa-
sis, and Transplantation Unit, for the excellent technical
tients with DGF. The colocalization of C4d with both C1q
assistance and Mariella Mastronardo for her editorial as-
and MBL in graft biopsies obtained from these patients
sistance and language revision of the manuscript.
indicated that both these pathway were activated onperitubular capillaries, within the interstitium, and on the
glomerular endothelium. Importantly, none of the patientsshowed histological signs of acute rejection, although we
1. Perico N, Cattaneo D, Sayegh MH, Remuzzi G: Delayed graft func-
cannot completely rule out the possibility that in some
tion in kidney transplantation. Lancet 2004, 364: 1814 –1827
patients C4d deposition might be due to the presence of
2. Gueler F, Gwinner W, Schwarz A, Haller H: Long-term effects of acute
circulating antibodies, undetectable by our standard
ischemia and reperfusion injury. Kidney Int 2004, 66: 523–527
methodology. However, it should be taken into consider-
3. Peeters P, Vanholder R: Therapeutic interventions favorably influenc-
ing delayed and slow graft function in kidney transplantation: mission
ation that none of the patients was treated for acute
impossible? Transplantation 2008, 85: S31⫺S37
rejection and all of them completely recovered graft func-
4. Sacks SH, Chowdhury P, Zhou W: Role of the complement system in
tion. In addition, our data are in agreement with previous
rejection. Curr Opin Immunol 2003, 15: 487– 492
demonstration of C4d or MBL deposits in human trans-
Li K, Sacks SH, Zhou W: The relative importance of local and
systemic complement production in ischaemia, transplantation and
planted kidney with early graft dysfunction39 or primary
other pathologies. Mol Immunol 2007, 44: 3866 –3874
non-function.40 To our knowledge, this is the first evi-
Ricklin D, Lambris JD: Complement-targeted therapeutics. Nature
dence of activation of classical and lectin pathway of
Biotechnol 2007, 25: 1265–1275
C1-Inhibitor in Renal I/R Injury
AJP April 2010, Vol. 176, No. 4
7. Walport MJ: Complement. First of two parts. N Engl J Med 2001, 344:
Porebski G, Hack CE, Verdonk R, Nuijens J, Levi M: Recombinant
human C1-inhibitor in the treatment of acute angioedema attacks.
8. Zhou W, Farrar CA, Abe K, Pratt JR, Marsh JE, Wang Y, Stahl GL,
Transfusion 2007, 47: 1028 –1032
Sacks SH: Predominant role for C5b-9 in renal ischemia/reperfusion
27. De Simoni MG, Storini C, Barba M, Catapano L, Arabia AM, Rossi E,
injury. J Clin Invest 2000, 105: 1363–1371
Bergamaschini L: Neuroprotection by complement (C1) inhibitor in
Thurman JM, Royer PA, Ljubanovic D, Dursun B, Lenderink AM,
mouse transient brain ischemia. J Cereb Blood Flow Metab 2003, 23:
Edelstein CL, Holers VM: Treatment with an inhibitory monoclonal
antibody to mouse factor B protects mice from induction of apoptosis
28. De Simoni MG, Rossi E, Storini C, Pizzimenti S, Echart C, Bergamaschini
and renal ischemia/reperfusion injury. J Am Soc Nephrol 2006, 17:
L: The powerful neuroprotective action of C1-inhibitor on brain isch-
emia-reperfusion injury does not require C1q. Am J Pathol 2004, 164:
Arumugam TV, Shiels IA, Strachan AJ, Abbenante G, Fairlie DP,
Taylor SM: A small molecule C5a receptor antagonist protects kid-
Storini C, Rossi E, Marrella V, Distaso M, Veerhuis R, Vergani C,
neys from ischemia/reperfusion injury in rats. Kidney Int 2003, 63:
Bergamaschini L, De Simoni MG: C1-inhibitor protects against brain
ischemia-reperfusion injury via inhibition of cell recruitment and in-
Mollnes TE, Kirschfink M: Strategies of therapeutic complement
flammation. Neurobiol Dis 2005, 19: 10 –17
inhibition. Mol Immunol 2006, 43: 107–121
30. de Zwaan C, Kleine AH, Diris JH, Glatz JF, Wellens HJ, Strengers PF,
12. Thorgersen EB, Ghebremariam YT, Thurman JM, Fung M, Nielsen
Tissing M, Hack CE, Dieijen-Visser MP, Hermens WT: Continuous
EW, Holers VM, Kotwal GJ, Mollnes TE: Candidate inhibitors of
48-h C1-inhibitor treatment, following reperfusion therapy, in patients
porcine complement. Mol Immunol 2007, 44: 1827–1834
with acute myocardial infarction. Eur Heart J 2002, 23: 1670 –1677
13. Davis AE, III, Mejia P, Lu F: Biological activities of C1 inhibitor. Mol
31. Heijnen BH, Straatsburg IH, Padilla ND, Van Mierlo GJ, Hack CE, Van
Immunol 2008, 45: 4057– 4063
Gulik TM: Inhibition of classical complement activation attenuates
14. Thurman JM: Triggers of inflammation after renal ischemia/reperfu-
liver ischaemia and reperfusion injury in a rat model. Clin Exp
sion. Clin Immunol 2007, 123: 7–13
Immunol 2006, 143: 15–23
15. Seelen MA, Roos A, Wieslander J, Mollnes TE, Sjoholm AG, Wurzner
32. Inderbitzin D, Beldi G, Avital I, Vinci G, Candinas D: Local and remote
R, Loos M, Tedesco F, Sim RB, Garred P, Alexopoulos E, Turner MW,
ischemia-reperfusion injury is mitigated in mice overexpressing
Daha MR: Functional analysis of the classical, alternative, and MBL
human C1 inhibitor. Eur Surg Res 2004, 36: 142–147
pathways of the complement system: standardization and validation
33. Bergamaschini L, Gobbo G, Gatti S, Caccamo L, Prato P, Maggioni
of a simple ELISA. J Immunol Methods 2005, 296: 187–198
M, Braidotti P, Di Stefano R, Fassati LR: Endothelial targeting with
16. Loverre A, Capobianco C, Stallone G, Infante B, Schena A, Ditonno
C1-inhibitor reduces complement activation in vitro and during ex
P, Palazzo S, Battaglia M, Crovace A, Castellano G, Ranieri E,
vivo reperfusion of pig liver. Clin Exp Immunol 2001, 126: 412– 420
Schena FP, Gesualdo L, Grandaliano G: Ischemia-reperfusion injury-
34. Sozzani S, Sallusto F, Luini W, Zhou D, Piemonti L, Allavena P, Van
induced abnormal dendritic cell traffic in the transplanted kidney
Damme J, Valitutti S, Lanzavecchia A, Mantovani A: Migration of
with delayed graft function. Kidney Int 2007, 72: 994 –1003
dendritic cells in response to formyl peptides, C5a, and a distinct set
17. Chatterjee PK: Novel pharmacological approaches to the treatment of
of chemokines. J Immunol 1995, 155: 3292–3295
renal ischemia-reperfusion injury: a comprehensive review. Naunyn
35. Nauta AJ, Daha MR, Tijsma O, van de WB, Tedesco F, Roos A: The
Schmiedebergs Arch Pharmacol 2007, 376: 1– 43
membrane attack complex of complement induces caspase activa-
18. Zhang M, Alicot EM, Chiu I, Li J, Verna N, Vorup-Jensen T, Kessler
tion and apoptosis. Eur J Immunol 2002, 32: 783–792
B, Shimaoka M, Chan R, Friend D, Mahmood U, Weissleder R, Moore
Pippin JW, Durvasula R, Petermann A, Hiromura K, Couser WG,
FD, Carroll MC: Identification of the target self-antigens in reperfu-
Shankland SJ: DNA damage is a novel response to sublytic com-
sion injury. J Exp Med 2006, 203: 141–152
plement C5b-9-induced injury in podocytes. J Clin Invest 2003, 111:
19. Zhang M, Takahashi K, Alicot EM, Vorup-Jensen T, Kessler B, Thiel
S, Jensenius JC, Ezekowitz RA, Moore FD, Carroll MC: Activation of
37. Nangaku M, Pippin J, Couser WG: Complement membrane attack
the lectin pathway by natural IgM in a model of ischemia/reperfusion
complex (C5b-9) mediates interstitial disease in experimental ne-
injury. J Immunol 2006, 177: 4727– 4734
phrotic syndrome. J Am Soc Nephrol 1999, 10: 2323–2331
20. Park P, Haas M, Cunningham PN, Bao L, Alexander JJ, Quigg RJ:
Hughes J, Nangaku M, Alpers CE, Shankland SJ, Couser WG,
Injury in renal ischemia-reperfusion is independent from immuno-
Johnson RJ: C5b-9 membrane attack complex mediates endothelial
globulins and T lymphocytes. Am J Physiol Renal Physiol 2002, 282:
cell apoptosis in experimental glomerulonephritis. Am J Physiol
Renal Physiol 2000, 278: F747⫺F757
21. Thurman JM, Ljubanovic D, Edelstein CL, Gilkeson GS, Holers VM: Lack
39. Feucht HE, Schneeberger H, Hillebrand G, Burkhardt K, Weiss M,
of a functional alternative complement pathway ameliorates ischemic
Riethmuller G, Land W, Albert E: Capillary deposition of C4d com-
acute renal failure in mice. J Immunol 2003, 170: 1517–1523
plement fragment and early renal graft loss. Kidney Int 1993, 43:
22. Collard CD, Montalto MC, Reenstra WR, Buras JA, Stahl GL: Endo-
thelial oxidative stress activates the lectin complement pathway: role
40. de Vries B, Walter SJ, Peutz-Kootstra CJ, Wolfs TG, van Heurn LW,
of cytokeratin 1. Am J Pathol 2001, 159: 1045–1054
Buurman WA: The mannose-binding lectin-pathway is involved in
Berger SP, Roos A, Mallat MJ, Fujita T, De Fijter JW, Daha MR:
complement activation in the course of renal ischemia-reperfusion
Association between mannose-binding lectin levels and graft sur-
injury. Am J Pathol 2004, 165: 1677–1688
vival in kidney transplantation. Am J Transplant 2005, 5: 1361–1366
41. Haas M, Ratner LE, Montgomery RA: C4d staining of perioperative
24. Nickeleit V, Zeiler M, Gudat F, Thiel G, Mihatsch MJ: Detection of the
renal transplant biopsies. Transplantation 2002, 74: 711–717
complement degradation product C4d in renal allografts: diagnostic
Sund S, Hovig T, Reisaeter AV, Scott H, Bentdal O, Mollnes TE:
and therapeutic implications. J Am Soc Nephrol 2002, 13: 242–251
Complement activation in early protocol kidney graft biopsies after
25. van Doorn MB, Burggraaf J, van Dam T, Eerenberg A, Levi M, Hack
living-donor transplantation. Transplantation 2003, 75: 1204 –1213
CE, Schoemaker RC, Cohen AF, Nuijens J: A phase I study of
43. Imai N, Nishi S, Alchi B, Ueno M, Fukase S, Arakawa M, Saito K,
recombinant human C1 inhibitor in asymptomatic patients with he-
Takahashi K, Gejyo F: Immunohistochemical evidence of activated
reditary angioedema. J Allergy Clin Immunol 2005, 116: 876 – 883
lectin pathway in kidney allografts with peritubular capillary C4d
Choi G, Soeters MR, Farkas H, Varga L, Obtulowicz K, Bilo B,
deposition. Nephrol Dial Transplant 2006, 21: 2589 –2595
Source: http://www.complement-system.eu/assets/upload/files/Complement%20Castellano%2010%20ED%20O.pdf
Social network wants to sequence your gut : Nature News Published online 8 September 2011 Nature doi:10.1038/news.2011.523 Social network wants to sequence your gut MyMicrobes project will gather DNA data from online community. Nicola Jones The non-profit programme MyMicrobes, launchedtoday, is inviting people to have their gut bacteriasequenced for about €1,500 (US$2,100). Acting asboth social network and DNA database, the websiteoffers a place for people to share diet tips, storiesand gastrointestinal woes with one another. Inexchange, researchers hope to gather a wealth ofdata about the bacteria living in people's guts.
GDP and Integrity of The Supply Chain Presented by: Karen S Ginsbury B.Pharm, MSc, MRPharmS IFF, October 2010 What are the Risks What are the Risks in GDP? • For a formal assessment need to use one of the tools in ICH Q9 / Annex 20: – HACCP– FMEA– Ishikawa (Fishbone) diagram + one of the • Will take you to a lot of the points already Every Picture Tells a Story







