Pro_nmeth983.indd
Protein complex expression by using
multigene baculoviral vectors
Daniel J Fitzgerald1, Philipp Berger2,3, Christiane Schaffitzel1, Kazuhiro Yamada1,
Timothy J Richmond1 & Imre Berger1
1ETH Zürich, Institut für Molekularbiologie und Biophysik, ETH-Hönggerberg, CH-8093 Zürich, Switzerland. 2ETH Zürich, Institut für
Zellbiologie, ETH-Hönggerberg, CH-8093 Zürich, Switzerland. 3Present address: Laboratory of Biomolecular Research, Molecular Cell Biology, Paul Scherrer Institute, CH-5232 Villigen PSI, Switzerland. Correspondence should be addressed to I.B. (
[email protected]).
Elucidation of the molecular basis of protein-interaction networks, in particular in higher eukaryotes, is
hampered by insufficient quantities of endogenous multiprotein complexes. Present recombinant expression
methods often require considerable investment in both labor and materials before multiprotein expression,
and after expression and biochemical analysis these methods do not provide flexibility for expressing an
http://ww altered multiprotein complex. To meet these demands, we have recently introduced MultiBac, a modular
roup baculovirus-based system specifically designed for eukaryotic multiprotein expression1. Here we describe
G new transfer vectors and a combination of DNA recombination–based methods, which further facilitate the
generation of multigene cassettes for protein coexpression (Fig. 1), thus providing a flexible platform for
generation of protein expression vectors and their rapid regeneration for revised expression studies. Genes
lishing
b encoding components of a multiprotein complex are inserted into a suite of compatible transfer vectors by
Pu homologous recombination. These progenitor constructs are then rapidly joined in the desired combination
by Cre-loxP–mediated in vitro plasmid fusion. Protocols for integration of the resulting multigene expression
cassettes into the MultiBac baculoviral genome are provided that rely on Tn7 transposition and/or Cre-loxP
Nature
6 reaction carried out in vivo in Escherichia coli cells tailored for this purpose. Detailed guidelines for multigene
virus generation and amplification, cell culture maintenance and protein production are provided, together
200
with data illustrating the simplicity and remarkable robustness of the present method for multiprotein
expression using a composite MultiBac baculoviral vector.
MATERIALS
REAGENTS
BD In-Fusion recombinase and buffers, (BD Bioscience)
L-arabinose (Gibco BRL)
Cre recombinase and 10× reaction buffer (NEB)
CellFECTIN (Invitrogen) or FuGENE (Roche)
Phusion high-fidelity DNA polymerase, 5× HF or 5× GC buffer
Template DNA (genomic, plasmid or cDNA)
(Finnzymes or NEB)
E. coli competent cells: TOP10, BW23473, DH10MultiBac
PCR primers (see Steps 3 and 4 for details)
Spodoptera frugiperda SF21 cells
AvrII,
PmeI,
SpeI (NEB),
Bsp68I (Fermentas) and
DpnI (NEB)
Vector DNA: pUCDM, pSPL, pFL, pKL, pFBDM, pKDM
QIAprep Spin Miniprep kit, QIAprep Gel Extraction kit,
Additional DNA constructs: pBADZHisCre, pUCDM-YFP
QIAprep PCR purification kit (Qiagen)T4 DNA ligase, 10× T4 DNA ligase buffer (NEB)
2×TY medium (1.6% tryptone, 1% yeast extract, 0.5% NaCl)
Thermocycler programmed with the desired protocols
TYE agar plates (1.0% tryptone, 0.5% yeast extract, 0.8% NaCl,
Equipment for agarose gel electrophoresis and SDS-PAGE
Shaker incubator (temperature-controlled, 27 °C)
Low-salt TYE medium (1.0% tryptone, 0.5% yeast extract, 0.5%
Erlenmeyer flasks (500 ml and 2 l)
NaCl) and agar plates (1.0% tryptone, 0.5% yeast extract, 0.5%
Fluorescence spectrophotometer and cuvettes (optional)
Electroporator and cuvettes
Sf-900 II SFM serum free medium (Gibco BRL)
Sterile hood with UV illumination
Antibiotics (see
Table 1)
6-well (35-mm diameter) tissue-culture plates
Isopropylthiogalactoside (IPTG; 1M)
4.5 ml CryoTube vials (Nunc)
15 ml and 50 ml Falcon tubes
PUBLISHED ONLINE 20 NOVEMBER 2006; DOI:10.1038/NMETH983
NATURE METHODS VOL.3 NO.12 DECEMBER 2006
1021
Multigene
1 Compile the DNA sequences encoding the components of the multiprotein complex of choice, and plan how
expression design
you will combine these genes into transfer vectors according to the schematics in
Figures 1 and
2.
and preparation of
target DNAs
2 All transfer vectors contain a multiplication module that can be used for combining expression cassettes
(
Fig. 2). If you decide to use the multiplication module, make sure the restriction enzymes used for
multiplication are not present in the genes you will be cloning.
▲
CRITICAL STEP
3 To insert DNA into the transfer vector of choice by seamless cloning using BD In-Fusion recombinase2,
design PCR primers that contain 20-30 bases annealing to the 5′ and 3′ end of the gene of interest. Add
a nonannealing 15–20-base 5′-tail to the primers containing DNA sequences that are homologous to the
sequences flanking the site of insertion into the transfer vector to facilitate In-Fusion recombination2.
Alternatively, insert encoding DNAs into transfer vectors by conventional cloning: amplify DNA fragments using PCR primers containing appropriate restriction enzyme sites for the chosen vector and 18–25 nucleotides complementary to the DNA sequence to be cloned. For both conventional and seamless cloning, if desired,
incorporate nucleotide sequences encoding for C- or N-terminal affinity tags into the primers.
4 For vector linearization by PCR, use primers annealing to the sequences flanking the site of insertion
(complementary to the 5′ tails of PCR primers in Step 3).
The plasmid used as vector also can be efficiently linearized by restriction digestion, optionally followed by gel extraction. Then nucleotide overhangs present at the restriction site have to be taken into consideration in primer
http://ww
design for the BD In-Fusion reaction as detailed by the manufacturer. In our hands, plasmid linearization by both methods (PCR amplification or restriction digestion) yield comparable results.
Prepare genes and vectors for multigene assembly (Steps 1–12)
Create ensemble of single gene-progenitor constructs
Combine genes into multigene expression cassettes (Steps 13–17)
Use
in vitro Cre-fusion and/or multiplication module (Fig. 2)
Integrate multigene transfer vectors into MultiBac bacmid (Steps 18–24)
pFL, pKL, pFBDM, pKDM
(antibiotic selection)
(blue/white selection)
DH10 MultiBacCre (Step 19) or DH10MultiBac (Step 23) cells
Occupy
loxP site first and Tn7 site next
Complex purificationand characterization;
Store bacterial clone
biochemical analysis
Isolate composite MultiBac bacmid (Steps 25, 26)
Transfect Sf21 cells
Generate production
500-ml shaker flask;
virus in 2-l shaker flask;
protein production in
6 ml initial virus (V
2-l shaker flasks
Figure 1 Outline of the method.
1022 VOL.3 NO.12 DECEMBER 2006
NATURE METHODS
5 Set up a 50-µl reaction for each DNA insert to be cloned and each vector to be linearized by PCR.
5× Phusion HF Reaction buffer
dNTPs (2.5 mM stock)
Template DNA (100 ng/µl)
5′ primer (10 µM stock)
3′ primer (10 µM stock)
Phusion polymerase (2 U/µl)
6 Amplify nucleic acid fragments using the following PCR program:
Cycle number
2 min/kb at 72 °C
30 s at 58 °C (decrease by 0.5 °C/cycle)
1 min/kb at 72 °C
http://ww
7 Digest PCR products for 4 h at 37 °C with 20 U of
DpnI enzyme.
DpnI can be added directly to the 50-µl
roup PCR. Supplement the reaction with 5
µl of 10× NEB buffer 4.
▲
CRITICAL STEP
Figure 2 Creating multigene transfer vectors. (
a) Vector maps (open arrows are expression cassettes; see also
Supplementary
Fig. 1). (
b) Multigene transfer vector assembly by
in vitro fusion of acceptor (pFL, pKL) and donor (pUCDM, pSPL) plasmid
derivatives by using Cre recombinase. Acceptor and donor plasmids all contain the
loxP imperfect inverted repeat (red circles).
Derivatives of one acceptor plasmid can be fused to one or two donor plasmids creating multigene plasmid dimers or trimers,
respectively. Expression cassettes in between the Tn7 transposition sequences (black triangles) are integrated into the MultiBac
baculovirus genome. (
c) Multigene assembly by using the multiplication module M present on all transfer vectors including also
pFBDM and pKDM (
Supplementary Fig. 1) following the procedure described1. Both approaches (
in vitro plasmid fusion and
multiplication via M) can also be used in conjunction for maximal flexibility in multigene vector assembly.
NATURE METHODS VOL.3 NO.12 DECEMBER 2006
1023
8 If PCR products appear to be sufficiently specific by analytical agarose gel electrophoresis, they can be used
directly (Step 9) following purification by QIAprep PCR purification kit. Alternatively, gel purify the desired
fragment using the QIAprep Gel Extraction kit.
Cloning of target
9 Set up BD In-Fusion reaction in 10 µl volume each:
DNAs into the
10× BD In-Fusion Reaction Buffer
PCR-amplified insert (200 ng/kb)
Linearized vector (100 ng/kb)
BD In-Fusion enzyme (diluted)
The BD In-Fusion enzyme stock concentration is 10×
; dilute to 1×
with BD In-Fusion dilution buffer and add 0.5
µ
l of this dilution to the reaction.
10 Incubate the reactions at 37 °C for 30 min and then at 50 °C for 20 min.
11 Use 1–5 µl of the BD In-Fusion reactions to transform chemically competent
E. coli cells (TOP10,
BW23474) and select for growth by plating on TYE agar plates containing the appropriate antibiotics for
selection (determined by the vector of choice; see
Table 1). Incubate overnight at 37 °C.
http://ww
▲
CRITICAL STEP
■
PAUSE POINT Plates can be stored for several weeks at 4 °C, and completed PCR and In-Fusion reactions
can be stored indefinitely at –20
12 Pick several clones, and isolate plasmid DNA using the QIAprep Spin Miniprep kit. Verify clones for each
target by restriction digestion, analytical PCR using the primers designed in Step 4 and/or by sequencing of
PCR product or plasmid DNA. Promoter and terminator elements can also be sequenced if vector linearization
by PCR was performed.
➨
TROUBLESHOOTING
▲
CRITICAL STEP
13 If both expression cassettes of a transfer vector are to contain an inserted gene each, repeat Steps 4–12
with a clone verified as in Step 12. Alternatively, if PCR steps are to be avoided, follow Steps 4–11 to generate constructs that contain one gene in one of the two expression cassettes of a transfer vector of choice and generate the dual expression vectors via the multiplication module and/or further convenient restriction sites present on the vector backbone.
Table 1 Features of MultiBac vectors
Replicon Host strain
Recombination and
Tn7L, Tn7R, multiplication Integration in MultiBac Tn7 site
Tn7L, Tn7R,
loxP,
Acceptor for plasmid fusions;
multiplication module M
integration in MultiBac Tn7 site
Tn7L, Tn7R, multiplication Integration in MultiBac Tn7 site
Tn7L, Tn7R,
loxP,
Acceptor for plasmid fusions;
multiplication module M
integration in MultiBac Tn7 site
Chloramphenicol R6Kγ
loxP, multiplication
Integration in MultiBac
loxP site;
donor for plasmid fusions
loxP, multiplication
Integration in MultiBac
loxP site;
donor for plasmid fusions
MultiBac bacmid Kanamycin
Tn7L, Tn7R,
loxP
Baculovirus receives Tn7 and
loxP
pBADZ-HisCre Zeomycin
Cre expression in DH10MultiBacCre
aYou may also use any other general laboratory cloning strain (
recA– endA– pir–).
1024 VOL.3 NO.12 DECEMBER 2006
NATURE METHODS
14 Design multigene expression cassette combinations following the schematic in
Figure 2. Plasmids
Creating
pSPL and pUCDM and their derivatives containing expression cassettes are called ‘donor plamids' (
Fig. 2
multigene
and
Supplementary Fig. 1). They contain a conditional origin of replication and a
loxP imperfect
expression
inverted repeat. Plasmids pFL and pKL and their derivatives are called 'acceptor plasmids' (
Fig. 2 and
Supplementary Fig. 1). Acceptor plasmids contain elements of the Tn7 transposon in addition to the
loxP sequence.
Donor plasmids containing expression cassettes can be fused via loxP to acceptor plasmids containing further
expression cassettes by in vitro Cre-Fusion (Fig. 2b). The resulting donor-acceptor fusions can then be integrated
into the MultiBac bacmid by Tn7 transposition. Donor plasmids can also be used directly for integration into the
MultiBac bacmid by in vivo Cre-loxP reaction (Step 18). Alternatively, multigene expression cassettes can also be
generated by using the multiplication module present on all vectors (Fig. 2c and Supplementary Methods) as
described previously1. Both approaches can also be used in combination.
15 Set up plasmid fusion reactions in 10-µl volume each.
10× Cre reaction buffer (NEB)
Donor plasmid A ( 500 ng)
Donor plasmid B ( 500 ng)
Acceptor plasmid ( 500 ng)
http://ww
If only one donor is fused to the acceptor, add 2 µ
l of ddH O instead of donor plasmid B.
roup
G 16 Use 1–5 µl of the Cre recombinase reaction products to transform competent
E. coli cells (for example,
TOP10) and select for growth by plating on TYE agar plates containing the appropriate resistance marker
lishing combinations (
Table 2). Incubate overnight at 37
Pu ▲
CRITICAL STEP
17 Pick several clones, isolate plasmid DNA using the QIAprep Spin Miniprep kit and verify clones for each
Nature
6 target by restriction digestion and/or sequencing.
200 ▲
CRITICAL STEP
■
PAUSE POINT Plasmids can be stored indefinitely at –20 °C.
18 Design a strategy to integrate multigene expression cassettes into the
loxP site and/or the Tn7 site of the
Preparing the
MultiBac bacmid (
Fig. 1). If it is desired to occupy the
loxP site with donors, this step must be carried out
composite
before Tn7 site occupation by fused donor-acceptors.
multigene
▲
CRITICAL STEP
19 Prepare DH10MultiBacCre cells by transforming the plasmid pBADZHisCre into competent DH10MultiBac
cells and selecting for growth on low salt TYE agar plates containing the appropriate antibiotics
dependent
(
Supplementary Methods), IPTG and X-gal for color selection. Incubate for >24 h at 37 °C with plates
integration of
wrapped in parafilm to avoid dehydration.
donor derivatives
For integration of acceptor plasmid derivatives or fused donor-acceptors via Tn7 transposition, use DH10MultiBac
into the MultiBac
cells instead (proceed to Steps 24–25).
▲
CRITICAL STEP
20 Pick a single blue colony and prepare electrocompetent DH10MultiBacCre cells that contain Cre
recombinase expressed as a result of L-arabinose induction (see
Supplementary Methods).
➨
TROUBLESHOOTING
▲
CRITICAL STEP
21 Incubate 10 ng of verified donor derivative for 15 min on ice with 50–100 µl electrocompetent
DH10MultiBacCre cells. After electroporation (200 Ω, 25 µF, 2.0 kV pulse), incubate cells for 8 h or overnight
at 37 °C and plate on TYE agar containing the selective antibiotics (
Table 2), IPTG and X-gal.
NATURE METHODS VOL.3 NO.12 DECEMBER 2006
1025
22 After 20–30 h incubation at 37 °C, blue colonies should appear. Inoculate blue colonies in appropriate
antibiotics and proceed to bacmid preparation for insect-cell infection (Step 25) or prepare electrocompetent
cells from clones for integration of an acceptor derivative by Tn7 transposition (
Fig. 1).
➨
TROUBLESHOOTING
▲
CRITICAL STEP
Tn7 transposition
23 Incubate 10 ng of verified acceptor derivative for 15 min on ice with 50–100 µl electrocompetent
of acceptor
DH10MultiBac cells. After electroporation (200 Ω, 25 µF, 2.0 kV pulse), incubate cells for 8 h or overnight at
derivatives into
37 °C and plate on TYE agar containing the selective antibiotics (
Table 2), IPTG and X-gal, and wrap plates in
the MultiBac
24 After 20–30 h incubation at 37 °C, white and blue colonies should appear. Pick white clones and proceed
to bacmid preparation for insect-cell infection (Step 25).
Preparation of
25 Incubate single clones containing composite MultiBac bacmid overnight at 37 °C in 2 ml of 2× TY liquid
bacmid for insect-
medium with the correct combination of antibiotics (
Table 2).
26 Prepare MultiBac bacmid DNA by alkaline lysis, for example, using solutions I, II and III of the QIAprep
Spin Miniprep kit following the protocol provided by the manufacturer. Precipitate the resulting supernatant
( 900 µl volume) with 800 µl isopropanol and wash pellet twice with 250 µl of 70% EtOH. Resuspend MultiBac bacmid DNA with 20 µl of filter-sterilized (0.22-µm) ddH O.
▲
CRITICAL STEP
http://ww
■
PAUSE POINT Bacmid DNA in isopropanol can be stored at –20 °C.
Table 2 Selection marker use
Host strain
pFL and pUCDM + pSPL
Amp., chl., spec.
pKL and pUCDM + pSPL
Kan., chl., spec.
loxP integration
MultiBac bacmid and
derivatives of pUCDM
MultiBac bacmid and
derivatives of pSPL
Kan. Tet. Spec.
Tn7 transposition
MultiBac bacmidd, and pFBDM DH10MultiBac
Amp., gent., tet.
or pFL derivativese MultiBac bacmidd and pKDM or pKL derivativese
Kan., gent., tet.
aConcentrations: ampicillin (amp.) 100 µg/ml; kanamycin (kan.) 50 µg/ml; chloramphenicol (chl.) 30 µg/ml; spectinomycin (spec.) 50 µg/ml; tetracycline (tet.) 10 µg/ml, gentamycin (gent.) 10 µg/ml. bOnly on agar plate: IPTG, 0.5 mM; X-gal, 500 µg/ml. c–, none. dWith or without donor derivatives integrated in
loxP site of bacmid. eIncluding double or triple donor-acceptor fusions.
1026 VOL.3 NO.12 DECEMBER 2006
NATURE METHODS
27 For every MultiBac bacmid DNA probe, seed 0.5 × 106 freshly diluted
S. frugiperda Sf21 cells in two wells
each of a 6-well tissue culture plate and incubate for 15 min at 27 °C (for cell maintenance see
Box 1).
Steps 27–34 should be performed in a sterile hood.
28 Combine in Eppendorf tubes 10 µl MultiBac bacmid DNA solution with 5 µl CellFECTIN transfection
reagent in 200 µl serum-free medium (Sf-900 II SFM). Incubate for 15 min at 27 °C.
▲
CRITICAL STEP
29 Add 1 ml of medium to the CellFECTIN-DNA suspension and use it to replace supernatant from seeded
cells. Seal 6-well plate with parafilm and incubate for 5 h at 27 °C. Then, aspirate off suspension and add
3 ml of fresh medium, and again seal with parafilm. Incubate at 27 °C.
30 After 48–60 h (at the latest), collect supernatant and store in a 15-ml Falcon tube. This is the initial virus
V ( 6 ml).
Optional: 3 ml fresh medium can be added per well if further progression of infection and/or protein production is to be monitored.
▲
CRITICAL STEP
31 In a 500-ml shaker flask, add 3 ml of V to 50 ml of freshly diluted cells at a density of 0.5
(cells/ml). Culture cells at 27 °C, with shaking at 80 r.p.m. Monitor cell count every 24 h. Split cells at 24 h
amplification and
intervals to below 1 × 106 until cell proliferation arrests. Simultaneously, swelling of the cells (about twofold)
should be observed. After an additional 48 h, collect the supernatant. This is generation 1 virus (V ).
(Pelleted
http://ww cells can be resuspended in fresh medium to monitor protein expression; Box 2.)
BOX 1 MAINTENANCE OF INSECT CELLS
Stock suspension cell culture. Maintain a liquid culture of Sf21 cells in 50 ml of SF-900 II SFM serum-free
medium at a cell count of 0.5–1 × 106 cells/ml in a 500-ml shaker flask at 27 °C, shaking at 80 r.p.m. Check cell count every 24 h using a light microscope. Flasks must be dedicated to insect-cell applications only, and cleaned and stored following standard cell culture–grade protocols.
Growing cells. For virus amplification, cells are seeded from the starter culture in a 2-l shaker flask in 100 ml
of medium at a cell count of 0.5 × 106 cells/ml. The volume should double to 200 ml (after 24 h) and to 400
ml (after 48 h). For protein expression, (Steps 31–33) cells are seeded from one 2-l shaker flask into a desired number of flasks. Fresh medium is supplemented in 24-h steps until a volume of 400 ml is reached per flask. For optimal aeration, cell count should not exceed 1.5 × 106 cells/ml throughout the procedure. If cells continue to double during protein production, it is advisable to split the cultures upon reaching a cell count >1.5 × 106 cells/ml by placing half of the volume into a fresh shaker flask and replenishing an equal amount of fresh medium.
Freezing and thawing cell stocks. A <2-week-old suspension cell culture maintained at 0.5–1 × 106 cells/
ml is centrifuged at 500
g for 5 min. The cell pellet is gently resuspended in fresh medium to a cell count of
3 × 107 cells/ml and DMSO is added to 10%. Aliquots (3 ml) in 4.5-ml cryo tubes are flash frozen in liquid
nitrogen and can be stored for up to several years.
For thawing, a 3-ml aliquot is incubated in a 37 °C water bath and immediately removed when thawed. The
cryo tube is then centrifuged at 500
g for 5 min. Medium is removed, and the cell pellet is gently resuspended in 2 ml of medium and transferred to a 50-ml Erlenmeyer flask. Fresh medium is added to achieve a cell count of 1 × 107 cells/ml. After approximately 4–6 d shaking at 80 r.p.m., cell division should begin and occur every 18 h. After 2 months, cells should divide every 24 h. Fresh stock suspension cultures should be started every 2 months.
Cell line used. We use Sf21 insect cells and Sf-900 II SFM for all steps involving cell culture. It has been
reported in the literature that certain cell lines are particularly suited for virus amplification, whereas
others are ideal for protein expression. Protein production and virus generation could be optimized by
testing various cell lines (Sf9, Hi-5), optionally supplemented with serum. But maintenance of one cell line
(Sf21) only for all purposes in serum-free medium reduces handling considerably and in our hands leads
to satisfactory results both for virus amplification, strorage and protein production in all cases tested. For
longer term storage of virus (>100 d), it can be advantageous to add fetal calf serum to 10% to reduce
virus aggregation and concomitant titer loss, or to freeze virus aliquots at –80 ºC.
NATURE METHODS VOL.3 NO.12 DECEMBER 2006
1027
BOX 2 MONITORING PROTEIN EXPRESSION
Expression analysis by SDS-PAGE. Protein expression can be monitored by SDS-PAGE at every step from initial
infection to protein expression by production virus. Protein production is often not manifest in cells used for
initial transfection on 6-well plates (unless fluorescent markers are used, see below). It is recommended to
monitor protein expression starting from cell proliferation arrest in cultures (
t = 0 h). Every 12 h (or 24 h),
1 ml sample is withdrawn from the liquid culture. Cells are pelleted for 1 min in an Eppendorf microcentrifuge
at 5,000 r.p.m. at 4 °C. Pellets are resuspended directly in 500 µl of SDS protein gel loading buffer with dye or,
alternatively, in 250 µl of a lysis buffer of choice (for example, PBS) supplemented by an equal volume of SDS
protein gel loading buffer. Short bursts (2–3 s) from a sonicator at low settings reduce viscosity of the sample.
SDS-containing samples are boiled for 10 min at 95 °C, centrifuged briefly at 14,000 r.p.m. in an Eppendorf
microcentrifuge and cooled on ice for 1 min before loading 5–10 µl on the gel.
Expression analysis using fluorescent markers. We have found it particularly useful to monitor heterologous
expression by following the specific signals of fluorescent proteins (for example, eYFP and/or eCFP)
cointegrated into the virus with the genes encoding for the proteins of choice. We have observed previously
that coexpression of fluorescent proteins did not reduce heterologous protein complex yield considerably,
even if large protein complexes of several hundred kilodaltons were expressed1. In the virus shown in
Figure
4, we had cointegrated eCFP in the Tn7 site and eYFP in the
loxP site of the MultiBac baculoviral genome.
From infected liquid culture, 1-ml sample ( 1 × 106 cells) was withdrawn and centrifuged at 5,000 r.p.m. in an
Eppendorf microcentrifuge for 1 min at 4 °C. Cell pellet was resuspended in 0.5 ml of PBS and treated with brief
bursts (2–3 s) from a sonicator at medium levels, followed by centrifugation at 14,000 r.p.m. in an Eppendorf
http://ww
microcentrifuge for 10 min. Expression of fluorescent proteins from both sites was conveniently monitored by fluorescence spectroscopy using the specific fluorescent signatures of eCFP and eYFP10. When both signals
reached a plateau, the cultures were collected and analyzed by protein SDS-PAGE. High-level fluorescent protein production was concomitant with maximal production of the respective protein complex.
32 In a 2-l shaker flask, add 5–10 ml V in 400 ml cell culture at 0.5 × 106 (cells/ml). Monitor cell count
every 24 h. Split cells to maintain a cell count below 1 × 106 until cell proliferation arrests. After an additional 48 h, collect the supernatant. This is generation 2 virus (V ).
Both V and V can be used for protein production. It is advisable to keep aliquots of V and V stored at 4
regeneration of production virus V , if needed.
■
PAUSE POINT All viruses (V , V and V ) can be stored for several months at 4 °C protected from light.
V and V are stored in sterile Falcon tubes. V production virus can be stored conveniently in emptied sterile
SF900 II SFM medium bottles.
33 For large-scale expression extending beyond 400 ml cell culture, multiply the setup outlined in Step 32
with further 2-l shaker flasks containing 400 ml of cell culture each. Use production virus V for infection.
It is advisable to monitor cell division in a pilot experiment with one shaker flask infected with 10–15 ml production virus V , as splitting cells below 1 ×
106 cells/ml if not enough virus was added is not convenient in this large scale
setup. The multiplicity of infection (MOI = ratio of virus particles per cultured cell) should be greater than 1. If cell proliferation continues in the pilot experiment upon virus addition beyond a cell count of 1.5 ×
106 (cells/ml), the volume of V virus added in a second pilot experiment should be increased to aim for a MOI > 1. Protein expression is
monitored throughout large scale protein production in at least one shaker as described (Box 2).
Step 6 There is unspecific or no PCR
The annealing temperature may have to be optimized. Alternatively,
annealing temperature can be successively decreased in the first five cycles down from 60 °C to 50 °C by 2 °C/step.
Steps 7, 12 There is high background of
Purify PCR reaction with QIAprep PCR Purification kit or, alternatively,
parental plasmids in spite of
DpnI
precipitate PCR reaction with NaOAc-EtOH after phenol-chloroform
digestion after transformation.
extraction. Resuspend DNA pellet in ddH O supplemented with
10× NEB buffer 4. Digest 10–12 h or overnight with 20–40 U
DpnI enzyme. A restriction enzyme which cleaves the parental plasmid but not the PCR product can be added to this reaction.
1028 VOL.3 NO.12 DECEMBER 2006
NATURE METHODS
Step 16 No transformants were obtained.
Repeat Step 15 with higher concentrations of donor(s) and acceptor plasmid DNA. In particular for triple donor-donor-acceptor fusions, efficiency of fusion is low and more colonies can be obtained by transforming into electrocompetent cells ( 109 cfu/µg).
Step 20 Cre is not expressed.
Make sure that low-salt agar and medium are used for plating and
preparing competent cells (see
Supplementary Methods). Use
freshly prepared L-arabinose solution for induction at OD
(not higher).
Step 22 No transformants were obtained.
Verify Cre expression level in DH10MultiBacCre cells by SDS-PAGE. If Cre is not highly expressed, prepare new batch of competent cells.
There are several white colonies among
Competent cell batch may be contaminated, since all colonies
blue colonies.
obtained must be blue at this step. Streak out DH10MultiBacCre
competent cells on 2× TYE plates containing X-gal, IPTG and
appropriate antibiotics (
Table 2) except for resistance marker
selecting for donor plasmid. All colonies obtained must be blue.
All colonies are white.
X-gal and/or IPTG in 2× TYE plates may be aged. Replate on agar containing fresh X-gal and IPTG.
Steps 31,32 Cells divide without
Initial V (Step 31) or V (Step 32) virus may have low titer. Repeat
infections with more virus added. Continue splitting cultures to below 1 × 106 each day for a total of 5–7 d and monitor cell count. If cell culture still divides after 7 d, repeat the initial transfection.
http://ww
Cell cultures stop dividing immediately
V or V was added at MOI > 1. This could lead to accumulation of
roup upon virus addition.
defective virus and loss of protein expression. Repeat infections with
less volume of respective virus.
No protein production is observed.
Cells may be aged. Repeat infections with cultures using freshly
prepared cells (
Box 1). Virus may have been overamplified. Start
again at Step 31 or 27 using less virus for infection. Safeguard that
cells divide once or several times upon virus addition (MOI << 1). Also for amplification take care to collect virus 48 h after infection. Make sure cell culture volumes do not exceed those indicated and
thereby restrict aeration.
200
CRITICAL STEPS
Step 2 The multiplication module can be used to create multigene expression cassettes (
Fig. 2c) and also
to conveniently generate dual expression cassettes from single gene progenitor plasmids. To be used for
multiplication, the restriction sites (
PmeI,
SpeI,
AvrII and/or
Bsp68I) have to be removed from the gene
ensemble, if present. Alternatively, for a complex containing three genes, for example, it may be best to use
two single-gene donors and one acceptor derivative joined by
in vitro Cre fusion.
Step 7 This step is necessary to remove methylated parental DNA and hemimethylated hybrids of one parental
and one PCR synthesized strand in the PCR reactions that would otherwise give rise to high background after
transformation. Alternatively, the linearization of the vector by PCR can be designed such that one or several
restriction enzyme sites that do not cleave the final construct are removed from the expression cassette by
placing the homology regions in the respective primers accordingly. Then, background can be suppressed by
restriction digestion with the enzyme of choice and/or
DpnI.
Step 11 The correct cell type has to be chosen for transforming BD In-Fusion reaction products. Donor
plasmids and derivatives contain a conditional origin of replication derived from R6Kγ and have to be
propagated in cell strains expressing the
pir gene such as BW23473 (
Table 1). Acceptor plasmids and
derivatives can be propagated in common laboratory bacterial strains (for example, TOP10 or DH5α).
Step 12 Phusion polymerase has the lowest currently reported error rate. Nonetheless, it is advisable to verify
the inserted gene as well as promoter and terminator regions of the constructs by sequencing. As several
copies of the same promoters and terminators are often used in a multiprotein expression experiment, it
is necessary to sequence individual promoter and terminator regions by ‘inside-out' gene-specific primers
annealing close to ( 100 bp) the 5′ and 3′ end of the genes.
NATURE METHODS VOL.3 NO.12 DECEMBER 2006
1029
Step 16 Owing to the conditional origin of replication, donor derivatives are not propagated in
pir– strains
(TOP10, DH5α and others). Cre-fusion reactions of donor and acceptor plasmids that are transformed into
strains lacking
pir give rise to double or triple fusions (
Supplementary Fig. 2) isolated based on resistance
marker combinations (
Table 2). We evaluated pairwise fusion efficiency by plating on agar plates containing
only one antibiotic selecting for the acceptor plasmid, or two antibiotics including the one provided by the
donor, thus selecting for fusions. We obtained 12–15% efficiency, which is consistent with previous reports. To
further facilitate multigene vector assembly, one can incubate both donors simultaneously with the acceptor
in a single reaction (
Fig. 2). Such fusions are isolated by plating on triple-resistant agar plates (
Table 2).
Although much less efficient than the pairwise reactions, we typically obtained 20–50 colonies from each
transformation reaction using 1 µg total DNA and our own CaCl competent cells (107 c.f.u./µg).
Step 17 Occasionally,
in vitro Cre-fusion can result in two identical donor derivatives fused to one acceptor
(see
Supplementary Fig. 2). Integration of such fusions into the MultiBac bacmid results in increased protein
production levels from the duplicated insert.
Step 18 It is best to occupy the
loxP site present on the MultiBac plasmid first if integrations both into the
Tn7 and
loxP sites are planned. From positive DH10MultiBacCre clones, electrocompetent cells containing the
recombinant bacmid are then prepared using the resistance markers indicated (
Table 2) following standard
protocols. Those competent cells are used for subsequent transformation with acceptor plasmid derivatives
which can contain (optionally) donor derivatives fused by
in vitro Cre reaction (
Fig. 1). In principle, it is
also possible to charge the Tn7 site in DH10MultiBac cells first (Step 23). In this case, however, plasmid
derivatives must be used that do not contain a
loxP site (pFBDM or pKDM rather then pFL or pKL, see
http://ww
Supplementary Fig. 1). Then, positive clones that contain recombinant MultiBac bacmid with genes inserted
into the Tn7 site can be used to generate competent cells expressing Cre recombinase (Steps 19, 20). These
competent cells are next transformed with donor derivatives.
Step 19 Antibiotic resistance by zeomycin is inhibited by high salt concentrations. Therefore, in both TYE
agar plates and in the culture used for preparing Cre-competent cells salt concentrations need to be adjusted
accordingly (see Materials and
Supplementary Methods).
Step 20 For successful integration of donor derivatives into the
loxP site of the MultiBac bacmid cells,
a strong expression of recombinant Cre recombinase must be observed by 15% SDS-PAGE analysis upon
induction with arabinose (
Supplementary Methods) and therefore should be monitored for each batch of
competent DH10MultiBacCre cells produced. Competent cells can be kept at –70 °C for months without a loss of
integration efficiency. Batches can be quality controlled by fusing a control plasmid containing a fluorescent
protein (for example, pUCDM-eYFP) and monitoring expression by fluorescence spectroscopy already at the
level of initial transfection (
Box 2).
Step 22 The blue color of the colonies indicates that the Tn7 transposition element, which is embedded in
a
lacZ gene, is intact and can be used for integration (
Fig. 1). All colonies on the plate should develop blue
color (see Troubleshooting for Step 22).
Step 26 It is very important to completely remove granular material that forms after cell lysis; otherwise
chromosomal DNA and other nucleic acids will contaminate the composite MultiBac bacmid. This can hinder
initial transfection (Step 29) since the ratio of total nucleic acid to lipofectant is crucial for success. We
recommend adding a second centrifugation step to the QIAprep protocol in fresh Eppendorf tubes. The
resulting supernatant should be clear of floating debris, and can be transferred to fresh Eppendorf tubes for
isopropanol precipitation.
Step 28 In addition to CellFECTIN, there are several other lipofectants which can be used as well for initial
transformation (for example, FuGENE). To date, we have carried out the majority of experiments in this
laboratory using CellFECTIN. Transfection with FuGENE in our hands works equally satisfactory and further
offers the advantage that the lipofectant suspension does not have to be removed after a 5-h incubation.
Step 30 (also 31,32,33) To maintain MOIs below 1 (ideally closer to 0.1) and thus to avoid drop of protein
expression levels through virus generations, it is mandatory to strictly follow the time course presented in
Steps 30–33. Initial virus has to be collected after 48–60 h, even if cells do not show clear signs of infection
and continue proliferating on the 6-well plate until confluency is reached (cells cover entire well). When using
shaker flasks, it is critical not to use cell culture volumes which are too large (500-ml flask, maximum 50 ml;
1030 VOL.3 NO.12 DECEMBER 2006
NATURE METHODS
2-l flask, maximum 400 ml). Also, it is particulary important to carefully control cell density and infect at each corresponding step at a cell count below 1 × 106 (cells/ml) (best between 0.5 × 106 and 0.7 × 106 (cells/ml)). Cell cultures during virus amplification have to be counted and split every 24 h to below 1 × 106 (cells/ml) until cell proliferation stops. Failure to strictly follow these guidelines may result in loss of protein expression due to lack of aeration if volumes and/or cell counts are too high. Upon infection, cells must still be proliferating during the first 24 h, otherwise the number of viruses per cell is above 1 (see Troubleshooting). Then, defective virus accumulates which can result in complete loss of heterologous protein expression.
COMMENTS
Multiprotein complexes with many subunits have become an intensive focus of current biological research.
Consequently, several multigene expression systems have been introduced for heterologous complex production.
Systems that have been used with success in E. coli include the pST44 polycistronic expression system3 and
the Duet system from Novagen4. For large eukaryotic complexes, we have recently introduced the MultiBac baculovirus system1. In contrast to expression in E. coli, proteins which are up to several hundred kilodaltons in size can be produced in insect cells infected with recombinant baculovirus, and the heterologous gene products generally are authentically processed and targeted to their native cellular component. Insect-cell expression using
baculoviral vectors is thus a convenient choice for production of large multiprotein complexes, which are the rule rather than the exception in eukaryotic cells5,6.
The vectors and protocols provided here were designed such that both the initial integration of
multiple genes into the baculoviral genome and the subsequent alteration of such gene combinations be as simple and rapid as possible. The modular use of the transfer vectors presented here, and the use of recombination-mediated fusion allows for multigene expression vectors to be reconstructed with ease
http://ww from progenitor donor and acceptor plasmids containing one or two genes each.
roup
A paramount issue for successful protein production using the baculoviral system is virus stability. In
G cell culture, it is often observed that deletion genotypes accumulate and eventually dominate the virus
population, ultimately resulting in virtually complete loss of heterologous expression7,8. Defective virus is preferentially produced in culture when infecting at high ratio of virus to cells or when collecting virus
lishing
b at times when the selective pressures from heterologous protein expression come to bear. Multigene
Pu baculoviral vectors based on the MultiBac bacmid can contain numerous repetitive elements (promoters,
terminators, recombination sequences) which may represent preferred sites for eliminating recombinant insert in vivo. We have analyzed amplified MultiBac viruses both by restriction mapping of purified viral
Nature
6 DNA (Fig. 3) and at single-cell resolution by immunofluorescence spectroscopy (Fig. 4), illustrating the
dangers of incorrect amplification of virus. Adherence to the protocol, in contrast, allows for amplification
200
of virus with constant protein production properties.
In principle, virus titer could be verified at every step for instance by classical methods such as plague
assay or end-point dilution9. The general protocols we provide here are not optimized in this respect, and it is clear that careful determination of individual virus titers and multiplicities of infection may be helpful for protein yield optimization. Our strategy relies on controlling MOI by dilution and splitting of cell cultures, thus avoiding lengthy analytical steps in the process from initial virus to protein production. Our setup is robust and standardized, and thus compatible with approaches in which numerous expression experiments are to be carried out in parallel in a time-saving way. There are many fermentors (for example, Wave Bioreactor, WAVE Biotech LLC; Biostat B, B.Braun Biotech int.) that can be used for large-scale cultivation of insect cells. In our hands, however, the use of ordinary laboratory shaker flasks as described in this protocol provided sufficient yield ( 1–20 mg/l depending on protein complex)
in most cases, even for structural biology applications.
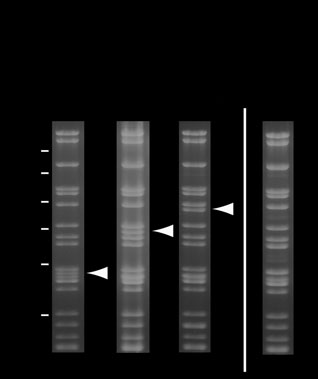
Figure 3 Stability of MultiBac baculoviral DNA analyzed by ethidium
bromide–stained 0.8% agarose gels. SalI-digested MultiBac viral DNA from
production virus (V ) gives the band pattern shown without recombinant
insert (lane 1), and with two and three genes, respectively, integrated into the Tn7 site (lanes 2 and 3). Position of bands corresponding to the
liberated DNA fragment containing the recombinant insert are marked with arrows (SalI does not cleave in the recombinant insert region). Virus
shown in lane 3 was in a separate experiment overamplified by serial passaging at high MOI leading to accumulation of viral DNA lacking the recombinant insert (lane 4).
NATURE METHODS VOL.3 NO.12 DECEMBER 2006 1031
M Sf21 V2 Pur.
http://ww
Figure 4 Protein complex expression by using MultiBac baculoviral vector. (a) Six genes were integrated into a MultiBac
baculovirus encoding a subcomplex of human TFIID (TAF5, 6 and 9) and fluorescent proteins eCFP (2 copies) and eYFP.
(b) Protein complex was purified from cell lysates infected with production virus V (SDS-PAGE). The arrow denotes bands
corresponding to fluorescent proteins. Virus V was generated as outlined. M, marker. (c) Infected cell cultures were assayed
for expression of YFP and TAF9 by immunofluorescence. Phalloidin (Phall.) stains both infected and uninfected cells. Cell
cultures infected with production virus (V ) produced the proteins in each cell assayed. Infection of cells with virus serial
passaged at MOI >> 1 revealed cells expressing either eYFP or TAF9 indicating that deletion virus had accumulated (OV, overamplified virus). Scale bars, 20 µm.
Note: Supplementary information is available on the Nature Methods website.
We thank Y. Hunziker for technical assistance, as well as D. Böhringer and C. Ostermeier for helpful comments. D.J.F. was a Human Frontier Science Program fellow, C.S. was a postdoctoral fellow of the Ernst Schering Research Foundation and P.B. was supported by the Swiss National Fund through a grant to U. Suter. T.J.R. acknowledges support from the Swiss National Fund through membership in the National Center of Competence in Research Structural Biology. I.B. acknowledges support from the Swiss National Fund.
COMPETING INTERESTS STATEMENT
The authors declare competing financial interests (see the Nature Methods website for details).
Published online at http://www.nature.com/naturemethods/
Reprints and permissions information is available online at http://npg.nature.com/reprintsandpermissions/
1. Berger, I., Fitzgerald, D.J. & Richmond, T.J. Baculovirus
et al. A protein interaction map of Drosophila
expression system for heterologous multiprotein complexes.
melanogaster. Science 302, 1727–1736 (2003).
Nat. Biotechnol. 22, 1583–1587 (2004).
7. Simon, O., Williams, T., Caballero, P. & Lopez-Ferber, M.
2. Benoit, R.M., Wilhelm, R.N., Scherer-Becker, D. & Ostermeier,
Dynamics of deletion genotypes in an experimental insect
C. An improved method for fast, robust, and seamless
virus population. Proc. R. Soc. Lond. B 273, 783–790
integration of DNA fragments into multiple plasmids. Protein
Expr. Pur. 45, 66–71 (2006).
8. Pijlman, G.B., de Vrij, J., van den End, F.J., Vlak, J.M. &
3. Tan, S., Kern, R.C. & Selleck, W. The pST44 polycistronic
Martens, D.E. Evaluation of baculovirus expression vectors
expression system for producing protein complexes in E.coli.
with enhanced stability in continuous cascaded insect-cell
Protein Expr. Pur. 40, 385–395 (2005).
bioreactors. Biotechnol. Bioeng. 87, 743–753 (2004).
4. Tolia, N.H. & Joshua-Tor, L. Strategies for protein coexpression
9. O'Reilly, D.R., Miller, L.K. & Luckov, V.A. eds. Baculovirus
in Escherichia coli. Nat. Methods 3, 55–64 (2006).
expression vectors. A laboratory manual. (Oxford University
et al. A comprehensive analysis of protein-protein
Press, New York, 1994).
interactions in Saccharomyces cerevisiae. Nature 403, 623–627
10. Patterson, G., Day, R.N. & Piston, D. Fluorescent protein
spectra. J. Cell Sci. 114, 837–838 (2001).
1032 VOL.3 NO.12 DECEMBER 2006 NATURE METHODS
Source: http://fs.teledos.gr:2206/%3ERESEARCH%20PUBLICATIONS/BIOLOGY/RNA%20and%20NOT%20DNA%20involved%20in%20diseases/Protein%20complex%20expression%20by%20using%20multigene%20baculoviral%20vectors.pdf
Characterization of extended-spectrum beta-lactamase-producing Salmonella enterica serotype Brunei and Heidelberg at the Hussein Dey hospital in Algiers (Algeria). Rachida Kermas, Abdelaziz Touati, Lucien Brasme, Elisabeth Le Magrex-Debar, Sadjia Mehrane, Fran¸cois-Xavier Weill, Christophe De Champs To cite this version: Rachida Kermas, Abdelaziz Touati, Lucien Brasme, Elisabeth Le Magrex-Debar, SadjiaMehrane, et al. Characterization of extended-spectrum beta-lactamase-producing Salmonellaenterica serotype Brunei and Heidelberg at the Hussein Dey hospital in Algiers (Alge-ria).
MONOGRAPHIE Pr ARIMIDEX® comprimés à 1 mg Inhibiteur de l'aromatase non stéroïdien AstraZeneca Canada Inc. Date de révision : 1004 Middlegate Road 27 avril 2011 Mississauga, Ontario L4Y 1M4 www.astrazeneca.ca Numéro de contrôle : 143918 ARIMIDEX® est une marque de commerce du groupe AstraZeneca.







