Research.cs.aalto.fi
Integrative and Personalized QSAR Analysis in
Cancer by Kernelized Bayesian Matrix
Muhammad Ammad-ud-din,∗,† Elisabeth Georgii,† Mehmet G¨
Laitinen,‡ Olli Kallioniemi,¶ Krister Wennerberg,¶ Antti Poso,‡,§ and Samuel
Helsinki Institute for Information Technology HIIT, Department of Information and
Computer Science, Aalto University, PO Box 15400, Espoo, 00076, Finland,
School of Pharmacy, Faculty of Health Sciences, University of Eastern Finland, PO Box
1627, Kuopio, 70211, Finland,
Institute for Molecular Medicine Finland FIMM, University of Helsinki, PO Box 20,
Helsinki, 00014, Finland,
Division of Molecular Oncology of Solid Tumors., Department of Internal Medicine 1,
University Hospital Tuebingen, Otfried Mueller-Strasse 10, 72076 Tuebingen, Germany,
Helsinki Institute for Information Technology HIIT, Department of Computer Science,
University of Helsinki, PO Box 68, Helsinki, 00014, Finland.
With data from recent large-scale drug sensitivity measurement campaigns, it is
now possible to build and test models predicting responses for more than one hundred
anti-cancer drugs against several hundreds of human cancer cell lines. Traditional quan-
titative structure-activity relationship (QSAR) approaches focus on small molecules in
searching for their structural properties predictive of the biological activity in a single
cell line or a single tissue type. We extend this line of research in two directions: (1) an
integrative QSAR approach, predicting the responses to new drugs for a panel of mul-
tiple known cancer cell lines simultaneously, and (2) a personalized QSAR approach,
predicting the responses to new drugs for new cancer cell lines. To solve the modeling
task, we apply a novel kernelized Bayesian matrix factorization method. For maxi-
mum applicability and predictive performance, the method optionally utilizes genomic
features of cell lines and target information of drugs in addition to chemical drug de-
scriptors. In a case study with 116 anti-cancer drugs and 650 cell lines we demonstrate
the usefulness of the method in several relevant prediction scenarios, differing in the
amount of available information, and analyze the importance of various types of drug
features for the response prediction. Furthermore, after predicting the missing values
of the data set, a complete global map of drug response is explored to assess treatment
potential and treatment range of therapeutically interesting anti-cancer drugs.
∗To whom correspondence should be addressed
†Helsinki Institute for Information Technology HIIT, Department of Information and Computer Science,
‡School of Pharmacy, Faculty of Health Sciences, University of Eastern Finland
¶Institute for Molecular Medicine Finland FIMM, University of Helsinki
§Division of Molecular Oncology of Solid Tumors., Department of Internal Medicine 1, University Hospital
kHelsinki Institute for Information Technology HIIT, Department of Computer Science, University of
Several recent large-scale high-throughput screening efforts provide drug sensitivity measure-
ments for a whole panel of human cancer cell lines and dozens of drugs. So far, these data
have been used in search for dependencies between genomic features and drug responses,
addressing the personalized medicine task.
The core computational problem of personalized medicine is the following: given genomic
features of the cell lines and their sensitivity measurements from an a priori fixed set of
drugs, predict sensitivity of a new cell line to these drugs. In other words the corresponding
computational models are devised to generalize across cell lines (Figure 1A). In personalized
medicine research, a variety of features have been studied for genomic characterization of
cell lines. These features include gene expression, copy number variation and mutational
status of the cell lines. For model learning, several statistical methods have been applied,
most commonly multivariate linear regression (using LASSO and elastic net regularizations)
and non-linear regression (e.g., neural networks, kernel methods).1–5
On the other hand, quantitative structure-activity relationship (QSAR) methods investigate
structural properties of drug molecules regarding their effects on the drug's biological activ-
ity, for an a priori fixed set of target cells.6 The resulting computational models can be used
for predicting the activity of drug compounds outside the data set, i.e., they have been de-
signed to generalize across drugs. (Figure 1B). Statistical methods for learning such models
are described in the next paragraph. Our work focuses on QSAR analysis of cancer drugs
and extends the traditional QSAR approach in two directions: (1) an integrative QSAR
approach: the responses to new drugs are predicted for a panel of representative cancer cell
lines simultaneously, and (2) a personalized QSAR approach: responses to both new and
established drugs are predicted for new cancer cell lines. The latter extension is developed
with the motivation to eventually be applied to primary cancer cell lines. Both extensions
are tested thoroughly using the data from the Sanger Genomics of Drug Sensitivity in Cancer
Two lines of research are crucial in the QSAR field: the development of suitable descrip-
tors capturing chemical and physical properties of drug molecules, and the development of
statistical methods to learn prediction models. The available descriptors range from 2D
fingerprints to spatial features and physiochemical properties.6,8,9 This paper contributes
to the second line, development of statistical methods, aiming at models that relate the
descriptors to biological activity. Previous work includes linear methods (e.g., multivariate
linear regression, partial least squares PLS, principal component regression PCR) as well
as non-linear methods (kernel methods, neural networks). In the specific problem of QSAR
analysis for cancer drugs, multivariate linear regression, PLS, PCR, kernel methods and neu-
ral network type methods have been widely applied. Linear approaches are most prominent
in QSAR analysis.6,8 Multivariate linear regression has been used in numerous studies,10–12
including analysis of anti-proliferative activity of compounds in human cancer cells.13 A ma-
jor challenge in the analysis is that structural descriptors can be highly correlated and the
number of descriptors is large, often exceeding the number of chemical compounds available
for training. There exist several ways to deal with these problems. A frequent solution
is to combine linear regression with feature selection techniques.14–16 In addition, sparsity
of linear regression models can be enforced by employing LASSO or elastic net regulariza-
tion.11 Another common strategy is principal component regression (PCR),6 which consists
of a two-step procedure. In the first step, the top principal components of the descriptor
data are computed to obtain a low-dimensional data representation by linear combinations
of the original descriptors. This representation is used in the second step as input to the
regression model. The main idea of partial least squares (PLS) is similar to that of PCR.
However, PLS additionally exploits the output values to construct the linear combinations
of the descriptors. PLS has been introduced to the QSAR field with Comparative Molecular
Field Analysis (CoMFA)17 and has been highly popular ever since.8,18 Also PLS methods for
multivariate output values have been used for QSAR studies18. In cancer analysis, PLS has
been applied to connect compound activity to genomic properties of cells19. Beyond the lin-
ear methods, nonlinear QSAR analysis has been studied. One approach is kernel regression.
For example, Yamanishi et al.20 predicted drug side effects from the chemical structure and
the target information of drugs using multiple kernel regression. An alternative approach
for nonlinear QSAR analysis are neural networks21. Sutherland et al.22 compared a neural
network-based QSAR model with other commonly known QSAR methods using benchmark
data sets. Their study reported that neural networks are equally or more predictive than PLS
models, when used with a basic set of 1D and 2D descriptors. However, with the availability
of high-dimensional data, the feasibility of simpler models such as traditional neural networks
depends on appropriate pre-selection of features to reduce the dimensionality of the data.
Recently, deep learning methods utilizing high-dimensional data have been studied for gen-
eral QSAR problems in chemoinformatics. Lusci et al.23 demonstrated a recursive approach
for modeling deep architectures for the prediction of molecular properties, and illustrate the
usefulness of their approach on the problem of predicting aqueous solubility. The method
proposed in this paper is also applicable to the general QSAR problem in chemoinformatics
and is fully equipped to deal with high dimensionality of the data.
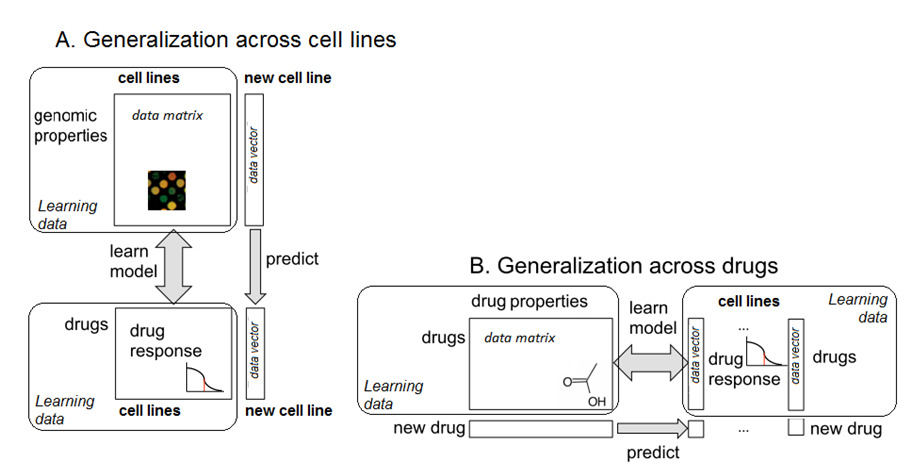
Figure 1: Two complementary approaches of exploiting high-throughput drug screening datafrom cancer cell lines: A. Personalized medicine approach (generalization across cell lines):given genomic properties of a cell line, predict the cell line's responses to a fixed set of drugs.
B. QSAR approach (generalization across drugs): given chemical and structural properties ofa drug, predict the response of this drug in a fixed set of cell lines. Each approach consists oftwo different steps. The first step is to learn a model that describes the relationship betweendrug responses and genomic (or chemical) properties. In the second step the model is usedto make predictions for new data with unknown response values. Methods to learn thesemodels are described in the main text. This paper extends the approach B, see Figure 2.
We present a novel matrix factorization method to address the task of QSAR analysis
in cancer. The method goes beyond the existing work in this application field in two main
aspects (Figure 2). First, multiple cancer cell lines are integrated into one model, instead
of building separate models for individual cell types13,24 or averaging across the cancer cell
lines25. This results in an integrative QSAR prediction, i.e., the columns of the drug response
matrix in Figure 2 are predicted together (not one by one independently), making it possible
to utilize their commonalities. Second, the model utilizes biological information not only
in the form of known targets as additional drug features, but also in the form of genomic
profiles of the cell lines. This allows us to also make predictions for untested drugs on new
cell lines (i.e., cell lines outside the training set). This can be viewed as personalized QSAR
prediction, which has not been addressed earlier and is a new research problem (Figure 2).
Technically, the method we propose for this task can be regarded as a type of general-
ized "recommender system", incorporating both side information on drugs (corresponding to
items) and side information on cell lines (corresponding to users). In previous work, matrix
factorization-based collaborative filtering, a popular approach in product recommendation
that does not use any side information, proved useful for predicting missing values of com-
pound activity in multiple cell lines26. Generalizing the matrix factorization to include side
information on both sides allows maximum flexibility in the various prediction scenarios. In
recommender system terminology, the integrative QSAR prediction corresponds to making
recommendations for the use of untested, cold-start compounds across the different cancer
types, whereas the personalized QSAR prediction considers cold-start compounds in combi-
nation with a cold-start cell line. Our recommender system is based on Kernelized Bayesian
Matrix Factorization method (KBMF), which was originally introduced for drug-protein in-
teraction analysis27 and has been extended to use multiple types of side information28. We
present a case study on drug response data from the Wellcome Trust Sanger Institute7. As
side information for drugs we use 1D, 2D, and 3D descriptors as well as protein targets. The
side information for cell lines includes gene expression, copy number variation, and muta-
multiple cell lines
new cell line
data matrices
(biological, chemical & structural)
properties (GE, CNV, Mut)
data matrices
drugs target -prints 1&2D Vsurf GRIND2
responses
Integrative QSAR
Figure 2: Extended QSAR analysis approach for cancer drug response prediction proposedin this work. Traditional QSAR analysis predicts efficacies of new cancer drugs from drugproperties (Figure 1B). The proposed approach additionally integrates side information onthe cell lines for which drug responses are predicted; here, the side information consists ofgenomic characteristics of the cell lines. With this approach, two new tasks can be performed:(1) integrative QSAR analysis: predicting drug efficacy for multiple cell lines simultaneouslyand (2) personalized QSAR analysis: predicting drug efficacy for a new cell line. In thefigure, each block represents a data matrix. The matrix of drug response values has one rowper drug and one column per cell line. The side information for drugs is given by a set ofmatrices, each of which represents the drugs as rows and drug features of a specific type ascolumns. Here, five drug feature types are considered: biological targets, fingerprints, 1&2Ddescriptors, Vsurf and GRIND/GRIND2. The contents of these data matrices are binary orquantitative values characterizing a specific drug with respect to the specific features. Theside information matrix for cell lines contains cell lines as columns and genomic features asrows (GE: gene expression, CNV: copy number variation and Mut: cancer gene mutations),only one block is shown to save space.
tion data. The method learns an importance weight for each data type and is implemented
as a Bayesian model, solving the extended QSAR problem by probabilistic modeling. The
kernel-based formulation allows for nonlinear relationships in the QSAR analysis.
Materials and Methods
Kernelized Bayesian Matrix Factorization (KBMF)
Drugs and cell lines are assumed to come from two domains X and Z, respectively. We
assume two samples of independent and identically distributed training instances from each
domain, denoted by X = {xi ∈ X }Nx and Z = {z
. For calculating similarities,
we use multiple kernel functions for each domain, namely, {kx,m : X × X → R}Px
{kz,n : Z × Z → R}Pz . The set of kernels may correspond to different notions of similarity
on the same feature representation or may use information coming from multiple feature
representations (i.e., views). The (i, j)th entry of the output matrix Y ∈
R x×Nz gives the
drug response measurement between drug xi and cell line zj.
Figure 3 illustrates the method we use; it is composed of three main parts included in
one unified model: (a) kernel-based nonlinear dimensionality reduction, (b) multiple kernel
learning, and (c) matrix factorization. First, we briefly explain each part and introduce the
notation. We then formulate a probabilistic model and derive a variational approximation
for learning and predictions.
Kernel-Based Nonlinear Dimensionality Reduction. This part performs feature
extraction using the input kernel matrices {K
and a common projection
, where R is the resulting subspace dimensionality. The projection gives
kernel-specific components {Gx,m = A>K
. The main idea is very similar to kernel
principal component analysis or kernel Fisher discriminant analysis, where the columns of
the projection matrix can be solved with eigen decompositions29. However, this solution
strategy is not possible for the more complex model formulated here.
Having a shared projection matrix across the kernels has two main implications: (i) The
number of model parameters is much lower than if we had a separate projection matrix for
each kernel, leading to more regularization. (ii) We can combine the kernels with multiple
kernel learning as explained in the following.
Figure 3: Flowchart of kernelized matrix factorization with multiple kernel learning. Fromeach of the side information data types for drugs illustrated in Figure 2, a pairwise similaritymatrix (kernel) between all the drugs in the training data set is computed (KX,1.KX,P ;
left border of image).
The model produces a low-dimensional representation of drugs
GX,1.GX,P , obtained from each kernel utilizing a common projection matrix A
based nonlinear dimensionality reduction, see text). A weighted combination of the matricesGX,1.GX,P parameterized by the weight vector e
X (one weight coefficient per kernel) yields
the composite component matrix HX (multiple kernel learning, see text). In the same way, acomposite component matrix HZ is obtained from kernels between cell lines, depicted on theright half of the figure. The output matrix Y (here, containing drug responses) is calculatedas a matrix product of HX and HZ (matrix factorization, see text). The grey shaded nodesare given by the training data, the green nodes are the parameters to be learned, and theblue nodes are the latent representations used by the model. See main text for more details.
Multiple Kernel Learning. This part is responsible for combining the kernel-specific
(i.e., view-specific) components linearly to obtain the composite components
x,mGx,m, where the kernel weights can take arbitrary values ex
multiple kernel learning property of this formulation can easily be seen from the following
where we need to have a shared projection matrix to obtain a valid linear combination of
the kernels.
Matrix Factorization. In this part, the low-dimensional representations of objects in
the unified subspace, namely, Hx and Hz, are used to calculate the output matrix Y = H>H
Figure 4: Graphical model of Kernelized Bayesian Matrix Factorization (KBMF) with mul-tiple kernel learning. The figure demonstrates the latent variables with their priors. Inparticular, Λ(.) denotes the matrices of priors for the entries of the projection matrices A(.).
η(.,.) represents vectors of priors for the kernel weights e(.,.). See text for more details ondistributional assumptions.
This corresponds to factorizing the outputs into two low-rank matrices.
Kernelized Bayesian Matrix Factorization (KBMF) is a probabilistic model formulated
to solve this prediction task. It has two key properties that enable us to perform efficient
inference: (i) The kernel-specific and composite components are modeled explicitly by intro-
ducing them as latent variables. (ii) Kernel weights are assumed to be normally distributed
without enforcing any constraints (e.g., non-negativity) on them.
Figure 4 shows the graphical model of KBMF with the latent variables and their pri-
ors. There are some additions to the notation described earlier: The N ×
matrices of priors for the entries of the projection matrices Ax and Az are denoted by Λx
z, respectively. The Px
1 vectors of priors for the kernel weights ex and
ez are denoted by ηx and ηz, respectively. The standard deviations for the kernel-specific
components, composite components, and target outputs are σg, σh, and σy, respectively;
these hyperparameters are not shown for clarity.
Next we specify the distributional assumptions of the model. The central assumptions
are normal distribution of the noise, and the dependencies shown in Figure 4; the other
assumptions are technical details made for analytical and computational convenience, and
can be replaced if needed. As the goal is to predict, these assumptions ultimately become
validated by prediction performances on the test data, see Results section. In the equations
matrices are denoted by capital letters, with the subscript x or z indicating the data domain
(drugs or cells, respectively); matrix entries are denoted by non-capital letters, with the row
index as superscript and the column index as the last subscript (i.e., ai
denotes the entry
at (row i, column s) of matrix Ax).
The distributional assumptions of the dimensionality reduction part are
∼ N (ai ; 0, (λi )−1)
x,s, kx,m,i ∼ N (gs
where N (·; µ, Σ) is the normal distribution with mean vector µ (here scalar) and covariance
matrix Σ (here scalar), and G(·; α, β) denotes the gamma distribution with shape parameter
α and scale parameter β. The multiple kernel learning part has the following distributional
ηx,m ∼ G(ηx,m; αη, βη)
ex,m ηx,m ∼ N (ex,m; 0, η−1 )
where kernel-level sparsity can be tuned by changing the hyperparameters (αη, βη). Setting
the gamma priors to induce sparsity, e.g., (αη, βη) = (0.001, 1000), produces results analo-
gous to using the 1-norm on the kernel weights, whereas using uninformative priors, e.g.,
(αη, βη) = (1, 1), resembles using the 2-norm. The matrix factorization part calculates the
target outputs using the inner products between the low-dimensional representations of the
x,i, hz,j ∼ N (yij
where I is the index set that contains the indices of observed entries in Y .
Exact inference for the model is intractable, and among the two readily available approx-
imative alternatives, Gibbs sampling and variational approximation, we choose the latter for
computational efficiency28. Variational methods optimize a lower bound on the marginal
likelihood, which involves a factorized approximation of the posterior, to find the joint pa-
The source code of the method is available as a Matlab package at http://research.
Benchmark QSAR Data sets
A total of eight standard QSAR data sets were used in the benchmark study22. The data sets
include (1) angiotensin converting enzyme inhibitors (ACE data set), (2) acetylcholinesterase
inhibitors (AchE), (3) benzodiazepine receptor ligands (BZR), (4) cyclooxygenase-2 in-
hibitors (COX2), (5) dihydrofolate reductase inhibitors (DHFR), (6) inhibitors of glycogen
phosphorylase b (GPB), (7) thermolysin inhibitors (THER), and (8) thrombin inhibitors
(THR). The details on these data sets including the number of compounds and the utilized
2.5D descriptors are presented in Table-S1 under the given acronyms (see Supporting In-
formation). The data sets were downloaded from the supplementary material provided by
Sutherland et al.22.
Drug Response Data
We used the data from the Genomics of Drug Sensitivity in Cancer project1,7 by Wellcome
Trust Sanger Institute (version release 2.0 July 2012) consisting of 138 drugs and a panel of
790 cancer cell lines. Drug sensitivity measurements were summarized by log-transformed
IC50 values (the drug concentration yielding 50% response, given as natural log of µM).
In addition, cell lines were characterized by a set of genomic features. We selected the
650 cell lines for which both drug response data and complete genomic characterization were
available. Furthermore, we focused on the 116 drugs for which SDF or MDL format (encoding
the chemical structure of the drugs) were available from the NCBI PubChem Repository,31
to be able to compute chemical drug descriptors. The resulting drug response matrix of 116
drugs by 650 cell lines has 75,400 entries, out of which 19,781 (26%) are missing. The used
data and value range were chosen to be consistent with earlier publications1,4.
As the KBMF model can incorporate multiple types of side information, we computed sev-
eral types of chemical descriptors for the drugs. First we computed PubChem finger print
descriptors using the PaDEL software32,33(v2.17, downloaded from the project website),
capturing occurrence of fragments in the 2D structure. In addition we calculated the 1&2D
descriptors available in PaDEL software, using default settings. The 1D descriptors are
summaries of compositional or constitutional molecular properties, e.g., atom count, bond
count, and molecular weight. The 2D descriptors encode different quantitative properties of
the topology32,33. Lastly we considered two types of 3D descriptors focusing on spatial and
physiochemical properties of the drugs, namely Vsurf and GRIND/GRIND2. The 2D struc-
tures were converted into 3D structures using the LigPrep module of the Schr¨
software package (Version 2.5, Schr¨
odinger, LLC, New York, NY, 2012). A few inorganic
substances were not successfully converted due to missing molecular parameters. A full set
of Vsurf descriptors was calculated from the 3D molecular database using Molecular Oper-
ating Environment software (MOE, Version 2012.10)34. Vsurf descriptors are generally used
to describe the molecular size and the shape of hydrophilic and hydrophobic regions. The
Pentacle software (version 1.0.6. Molecular Discovery Ltd. 215 Marsh Road, Pinner, Mid-
dlesex, UK) was applied to calculate GRIND and GRIND2 descriptors35–37. First, 3D maps
of interaction energies between the molecule and chemical probes were calculated. Second,
these 3D interaction maps were further converted into GRIND and GRIND2 descriptors,
which do not require alignment of compounds. In a preprocessing step, we removed all fea-
tures with constant values across all drugs, obtaining the final set of features for each type
listed in Table S-3 (Supporting Information). In addition to these chemical and structural
descriptors, we obtained drug target information from the Genomics of Drug Sensitivity in
Cancer project7 and encoded it as a binary drug versus target matrix. In addition to indi-
vidual target proteins, target families and effector pathways were included. For each type of
drug features, a kernel matrix was computed, containing pairwise similarities of drugs with
respect to the specific feature type. For the binary features (targets and finger prints), the
Jaccard coefficient was used; for all other feature types a Gaussian kernel was computed.
Cell Line Features
Three types of genomic profiles were included in the Genomics of Drug Sensitivity in Cancer
project1 to characterize the cell lines: gene expression, copy number and mutation profiles.
Gene expression profiles quantize the transcript levels of genes, whereas copy number pro-
files measure the amplification or deletion of genes in the DNA, and mutation profiles state
changes in the sequence of a gene. While the gene expression and copy number profiles
are genome-wide, the mutation data focus on cancer gene mutations and relate to somatic
mutations frequently occurring in tumours. In the Genomics of Drug Sensitivity in Can-
cer project1, the gene expression measurements were made using HT-HGU122A Affymetrix
whole genome array, copy number variants were obtained through SNP6.0 microarrays, and
cancer gene mutations were determined using the capillary sequencing technique. We in-
cluded all three data types in our analysis, using the same data as the elastic net analysis in
the pilot study, accessible from Genomics of Drug Sensitivity in Cancer project's website7.
Analogously to the preprocessing of drug features, we removed the features with constant
values across all cell lines. The number of features for each type is given in Supporting
Information, Table S-4. To obtain a kernel representation, the Jaccard similarity coefficient
was used for mutation data and Gaussian kernels for gene expression and copy number data.
Results and Discussion
Performance Evaluation on Benchmark QSAR Data sets
We started by verifying that in the standard QSAR task, where the new approaches do not
have a competitive advantage, the performance of the new KBMF method was comparable
to state-of-the-art QSAR methods. We evaluated the methods using the eight benchmark
data sets with training data, test data and descriptors provided by Sutherland et al.22 in
their comparison study. During training, a nested cross-validation experiment was performed
to select the optimal number of components for KBMF, which was subsequently used for
predictions on the test set. Details can be found in Supporting Information (Text, Table
S-2). Table 1 lists the performance on the test sets, using the same evaluation criteria
as reported in the earlier benchmark experiment22, coefficient of determination (R2 ) and
standard error of predictions (stest). Additionally, we provide error statistics for KBMF.
Regarding the summary performance measures, the KBMF method achieves the best results
among all methods in five out of eight data sets, showing that KBMF performance matches
the state of the art. Like the other methods, KBMF achieves the best prediction performance
for the data sets ACE and DFHR, and in both cases it has better performance values than
the other methods. For the BZR data set, KBMF and GFA-l have comparable performance,
following NN and NN-ens. Sutherland et al.22 reported the presence of outlier compounds in
this data set. Removing outliers as described there, the R2
test improved to 0.49 and
0.67, respectively, outperforming NN and NN-ens. In the THER data set, the performance
of KBMF is comparable to PLS but lower than for the remaining methods. One potential
reason for the better performance of the other methods on this specific data set is that
they pre-select relevant features. Feature selection is necessary for the feasibility of GFA
and traditional NN type of models, whereas deep and recursive NN, PLS and KBMF can
operate on the full data with high dimensionality. It is plausible that performance of KBMF
could also improve with suitable feature selection but this investigation is left to future work.
Table 1: KBMF test set performance comparison against state-of-the-art QSAR methodsusing benchmark QSAR data sets. Performance indicators of Partial Least Squares (PLS),Genetics Function Approximation (GFA), GFA with linear terms (GFA-l), GFA ensemblemodels with linear terms (GFA-l-ens), GFA with nonlinear terms (GFA-nl), GFA ensemblemodels with nonlinear terms (GFA-nl-ens), PLS models with descriptor selection via GFA(GPLS), ensemble of PLS models with descriptor selection via GFA (GPLS-ens), NeuralNetworks (NN), and ensemble of Neural Network models (NN-ens) were taken from thework by Sutherland et al.22 their Table 5. The KBMF method used the same training andtest sets as the others. The best result for each data set is marked in bold. Additionally, weprovide the pooled mean squared error with standard error of the mean (SEM) and standarddeviation (in brackets). For the comparison methods detailed statistics are not available andwe computed only MSE from the stest value.
1.12±0.42 (1.58)
1.16±0.26 (1.63)
0.68±0.17 (1.18)
1.13±0.24 (2.34)
0.84±0.11 (1.19)
0.86±0.26 (1.22)
4.97±0.45 (2.24)
0.80±0.24 (1.27)
Integrative QSAR Prediction of Drug Responses across Multiple
Cancer Cell lines
We next present an integrative QSAR analysis on the drug response data from the "Genomics
of Drug Sensitivity in Cancer" (GDSC) project7. Unlike the benchmark QSAR applications,
which have a single output variable, the drug sensitivity study investigates the efficacy of
compounds across a set of different cell lines and cancer types. Rather than looking at
each cell line one by one, a natural extension of QSAR analysis considers all cell lines
simultaneously in one unified model, which is implemented by the KBMF method. We
utilized 1D, 2D, and 3D drug descriptors for predicting the response of left-out drugs (eight-
fold cross validation). The number of components in the KBMF method was fixed to 45 for all
experiments on the GDSC data, which corresponds to a dimensionality reduction of about
sixty percent relative to the smaller dimension of the response matrix (here, the number
of drugs). Three different evaluation measures are reported on the test data: (1) the mean
squared error (MSE), (2) the coefficient of determination R2 and (3) Pearson correlation RP .
We complement the MSE value with estimates of standard errors and standard deviations.
The cross-validation statistics for these evaluation measures can be found in the Supporting
Information (Table S-5).
Improved Performance by Incorporating Biological Information
In addition to the 1D, 2D, and 3D chemical and structural descriptors of drugs, we incorpo-
rated biological features into the model, in the form of (1) target information for the drugs
and (2) genomic properties of cell lines, both provided by the "Genomics of Drug Sensitivity
in Cancer" project7. Neither of the two types of biological feature information is used in
traditional QSAR approaches. We note that integrating the target information is easy in all
QSAR approaches. However, simultaneous integration of genomic information has not been
reported with any state-of-the-art QSAR methods for this prediction task. Our analysis
shows that providing biological information to the model increases the prediction perfor-
mance (see Table 2, and Supporting Information Table S-5 for cross-validation statistics).
The predictive performance of the method KBMFDD+T R+CF is significantly higher than the
other methods with a p-value < 0.01 (Supporting Information Tables S6 and S7). In terms
of absolute performance, R2 and Rp are at moderate levels but significantly better than
Table 2: Predicting responses to new drugs for a panel of known cell lines, applying KBMFwith different feature sets. MSE values with standard error of the mean (SEM) and standarddeviations (in brackets) are shown. The subscript denotes the combination of feature typesused in each of the experiments: DD=Drug Descriptors, TR=Targets, CF=Cell Features.
The best result for each performance measure is marked in bold. The results indicate thatbiological features improve drug response prediction. Biological features can be either targetinformation or genomic measurement data (neither are used in traditional QSAR analysis,but the former is easy to integrate into existing QSAR approaches).
0.83±0.0053 (1.24)
0.72±0.0045 (1.07)
0.80±0.0051 (1.21)
0.69±0.0043 (1.01)
Baseline1 (cell line-wise mean prediction)
1.03±0.0061 (1.50)
Baseline2 (overall mean prediction)
1.04±0.0064 (1.52)
baseline methods that take the mean of the training data as the prediction for a new drug
(Baseline1: cell line-specific mean, Baseline2: overall mean). Therefore, we can conclude
that KBMF is appropriate to address the novel and challenging task of predicting responses
to new drugs for a panel of cell lines.
Relative Importance of Individual Drug Descriptor Types for Predicting Drug
We investigated the contribution of each drug feature type to the task of predicting responses
for test drugs left out from the training data. KBMF learns relevance weights ex,m for
the different feature types. Figure 5 shows the average relevance weights across the eight
models from cross-validation. The target information obtained the largest weight and the
GRIND/GRIND2 descriptors the lowest weight. Remarkably, the relative ranking of feature
types is conserved in all eight models (see Supporting Information, Figure S-1), indicating
robustness of this relevance ordering. To analyze whether already a subset of feature types
gives sufficient predictive power, we tested the performance of different combinations, adding
feature types in the order of their learned weights (Table 3). While target information alone
already achieves a considerable fraction of the overall performance, further performance gains
are achieved by adding the other feature types, with the best result being obtained by the
combination of all feature types (see Table 3, and Supporting Information Table S-8 for
cross-validation statistics). Even though, the differences in performances are quite small,
the predictive performance obtained by the combination of all feature types is found to be
significantly higher than the others with a p-value < 0.01 (Supporting Information Tables
S-9 and S-10).
Table 3: Prediction results with different combinations of feature types. MSE values withstandard error of the mean (SEM) and standard deviations (in brackets) are shown. Inte-gration of all drug feature types gives the best result.
0.71±0.0048 (1.13)
Targets and fingerprints
0.73±0.0045 (1.07)
Targets, fingerprints and 1&2D
0.72±0.0045 (1.06)
Targets, fingerprints, 1&2D and Vsurf
0.72±0.0045 (1.06)
Targets, fingerprints, 1&2D, Vsurf and GRIND/GRIND2
0.69±0.0043 (1.01)
Figure 5: Relevance weights learned by the KBMF method for the different drug featuretypes, averaged across the eight models from the cross-validation experiment. The rankingof feature types is conserved in the individual models (Figure S-1 Supporting Information).
A Global Map of Drug Responses in Cancer
We will next discuss the global map of drug response in cancer, produced by predicting miss-
ing values from the data. In the observed data, 26% of the measurements are missing, and
we used KBMF to predict these missing values. We will begin by validating the performance
of the method on missing value prediction, and then, for more detailed analysis we pick a set
of therapeutically interesting drugs with reliable predictions. Therapeutic interestingness of
the drugs is judged based on the selectivity of responses, and reliability was estimated by
cross-validation on the existing measurements.
Performance on Missing Value Prediction
Up to now in this paper, we have investigated performances in predicting responses to
previously unseen drugs. In practice, also the following prediction task is relevant: given
response measurements for a drug in a subset of cell lines, predict drug responses on the
remaining cell lines. In the context of recommender systems, this is called a warm-start
prediction task because it can use partially known response values. We tested two warm-
start prediction scenarios. In the blockwise approach, the same set of cell lines is missing for
all drugs in the set of test drugs, whereas in the entrywise approach scattered drug-cell line
combinations are predicted (Table 4, and Supporting Information Table S-11). In both cases
the prediction performance is much better than when response values to the test drug are
unknown for all cell lines (cf. Table 2). That is, the method is able to exploit the additional
available information to improve predictions. For our further analysis in the next section,
we consider on the drugs based on the reliability of their predictions, as measured by M SE,
focusing on the drugs with lower-than-average M SE in the entrywise prediction benchmark
Table 4: Performance on missing entry prediction (a warm-start prediction task) using theKBMF method. Two different variants of the task are evaluated, predicting entries of amissing block (blockwise) and predicting scattered entries (entrywise). MSE values withstandard error of the mean (SEM) and standard deviations (in brackets) are shown. Com-paring to Table 2, note the additional performance gain when including known sensitivitymeasurements of the test drug on some cell lines into the model (and predicting for theremaining cell lines).
0.26±0.0020 (0.47)
0.21±0.0016 (0.37)
Insights from Novel Predictions
Using the method validated in the previous subsection, we trained a model on all available
data and used it to predict the missing responses in the Sanger data set. We will next discuss
insights from these predictions, for the subset of drugs whose predictions are reliable (chosen
as discussed in the previous subsection), exhibit tissue-selectivity of response (as evaluated
by ANOVA), and being new findings (based on newly predicted values instead of existing
data). In practice, we selected drugs with at least 20 newly predicted values, grouped cell
lines based on their tissue origin and measured selectivity of response by a one-way ANOVA
test for each drug against these groups, retaining drugs with p-value < 0.05, and analyzing
their tissue-specific effects. Effects are calculated as the difference between group mean and
grand mean (effect=group mean − grand mean); smaller values correspond to sensitive tissue
types, larger values to resistant tissue types.
Figure 6 summarizes the tissue-specific effects of the selected drugs. In the figure, the drugs
have been categorized into three groups of response patterns. Group 1 consists of drugs
that are more effective on blood cancer than on lung cancer. This pattern is exhibited by
nutlin-3a, palbociclib, nilotinib, SB590885, olaparib, veliparib and bosutinib. According to
the database at National Cancer Institute (NCI)38, bosutinib has been approved for a certain
type of blood cancer. Group 2 shows the reverse pattern of group 1, being more effective in
lung than in blood cancer, and includes the drugs BI-D1870, ZM-447439, vorinostat, PD-
173074 and BX-795. Group 3 contains drugs that are effective in more than one cancer
types, namely lapatinib, saracatinib, BMS-509744, WZ-1-84 and crizotinib. Lapatinib is
approved for breast cancers but additionally seems to have strong response in upper aerodi-
gestive cancers. As another example, crizotinib is approved for lung cancers but appears to
have some selective activities in CNS, upper aerodigestive, and GI tract cancers. Supporting
Information Figures S-2 and S-3 shows the heatmaps of the predicted response values for
these selected drugs across cell lines from the seven major tissue types.
Finally we discuss properties of the full response data, containing both the observed data and
the newly predicted values for unobserved drug-cell line combinations (focusing on the seven
major tissue types from Figure 6), giving a global view on treatment potential as shown in
the Figure 7 (and Supporting Information Figure S-4). We observe that the drugs camp-
tothecin, doxorubicin, docetaxel, paclitaxel, vinorelbine, vinblastine, epothilone-B, borte-
zomib and thapsigargin show efficacy across many cell lines, spanning many cancer tissue
types. This is in line with the known evidence about these drugs in NCI database, as many
of them have been approved for several types of cancers and, importantly, they are gen-
eral cytotoxic or anti-mitotic drugs and therefore affect many, if not all, cancer cell lines
(in particular doxorubicin, docetaxel, paclitaxel, vinblastine, and vinorelbine). On the other
hand, all cancer types are resistant to the drugs cyclopamine, GDC-0449, LFM-A13, AICAR,
metformin, WZ-1-84, NSC-87877, KIN001-135, KU-55933, lenalidomide, ABT-888, DMOG,
Figure 6: Tissue effects from one-way ANOVA for a set of potentially therapeutic drugs.
Each column shows all the tissue effects for one drug. The color represents the cancer tissuetype. The size of the circle denotes the number of missing values in the original data, forwhich response values have now been computationally predicted. Effects are calculated asthe difference between group mean and grand mean; smaller values correspond to sensitivetissue types, larger values to resistant tissue types. Three visually clear interesting groupsof drugs have been denoted by Groups 1, 2 and 3.
JNK-Inhibitor-VIII, and BIRB-0796. Some aspects of drug activity become only visible in
the map that includes the newly predicted values; they were not evident from the observed
data only. One example is the finding that the strongest response of the drug AZ628 is
achieved in a large set of skin cancer cell lines. The efficacy of this drug on melanoma, a
type of skin cancer, is known from the literature39. Another interesting observation from the
global map is that dasatinib and WH-4-023 show in large parts similar response patterns,
and strong efficacy for a small number of blood cell lines, which is consistent with the NCI
annotation of dasatinib. The two drugs have some structural similarities on the 2D level
and share certain targets (SRC family and ABL). Bosutinib also hits these targets, and has
similar but weaker response patterns. Mitomycin-C exhibits activity spanning various tissue
types and has been approved for GI tract and pancreatic cancers.
The fully predicted drug response map can be used as a resource for generating new hypothe-
ses on cancer drug treatment and repositioning of drugs, suggesting targeted experiments for
further studies. For instance, the reddish vertical stripes seen for several drugs suggest wide
effective responses and broad applicability, whereas small red patches exhibit more localized
responses and applicability for specific types of cancer (e.g., blood).
AICAR or − ZHZHH
fSKHLL SZdKKF AZFdZ
Bosutinib albociclib NU
− AZDFLZd Lapatinib Er
GSKdFKKFdA BA −
Figure 7: A global drug response map showing responses of 482 cancer cell lines from seventissue types to 116 drugs. The map was compiled from predicted missing data(25.4 %) andobserved data. The annotation bar (left) represents the cell line tissue type. The color key(top) shows the range of the logIC50 responses.
The Novel Task of Personalized QSAR Analysis
Finally, we address the novel task of predicting responses to new drugs for new cell lines.
This can be considered as personalized QSAR analysis, i.e., finding suitable drugs for new
cancer cell line(s) or patient(s) (see Figure 2). When predicting values for combinations
of held-out drugs and held-out cell lines, KBMF achieved M SE, R2, and RP values of
0.78±0.012(0.92), 0.20 and 0.52, respectively. Since this prediction task is very challenging,
the performance is lower than for the task where the set of cell lines is known beforehand
(cf. Table 2). However, when the cell line is new the approach is significantly better (with
a p-value < 0.01; Supporting Information Table S-13 and S-14) than what is available as
baseline; predicting the mean of the training data yields an MSE of 1.08±0.015(1.24).
As this is a new task, we cannot compare to existing approaches and we quantify the quality
of personalized QSAR predictions by using the easier prediction tasks above as a yardstick.
We chose the missing value predictions (i.e., warm start) as the reference and checked the
fraction of residuals (observed value minus predicted value) falling into percentiles around
the mean of the warm start distribution (Figure 8). About 60% of the personalized QSAR
prediction residuals and 55% of the baseline residuals are covered by the whole range of
missing value prediction residuals, whereas at the 60% quantile, 20% of personalized QSAR
prediction residuals and 10% of baseline residuals are covered. In summary, while a larger
amount of available information increases the prediction accuracy, the proposed KBMF
method outperforms the available baseline in the challenging de-novo prediction task.
To further support our findings, we additionally compared the distribution of entry-wise
residuals with the residual distributions from other prediction tasks (Supporting Information
Figure S-5) . The missing value prediction task where both the cell line and the drug
have been observed earlier (but not in combination) has the narrowest residual distribution,
peaking at zero. The other prediction tasks yield much more wide-spread distributions,
differing less from each other than from the missing value prediction scenario.
These results show that it is possible to tackle the new personalized QSAR prediction task
with machine learning approaches. It would be possible to adapt some of the other recent
methods4 to the task as well, and assessing the relative merits and clinical impact will
naturally need further studies.
Personalized QSAR
Figure 8: Percentage of prediction residuals (y-axis) falling into percentiles around the meanof missing value prediction residuals (x-axis). The bars around the points denote the standarddeviation at a particular percentile. Personalized QSAR (blue color): predicting responsesto new drugs and new cell lines using KBMF approach. Baseline (red color); predictingthe mean of the training data. The KBMF method outperforms the baseline approach inthe challenging task of predicting personalized QSAR responses. Full residual distributionsof the baseline, the personalized QSAR prediction and the missing value prediction can befound in Supporting Information Figure S-5.
We presented an extended QSAR analysis approach using Kernelized Bayesian Matrix Fac-
torization (KBMF). Our two main conceptual contributions are: (i) an integrative QSAR
approach predicting responses to new drugs for a panel of known cell lines and (ii) a per-
sonalized QSAR approach predicting responses to new drugs for new cell lines. Earlier
methods have not been available for the personalized QSAR task and in the simpler tasks
our experiments have shown that KBMF was at least as good as the existing methods. A
case study on the large Sanger cancer drug response data set demonstrated the feasibility
of these prediction scenarios and showed that the use of multiple side information sources
for both drugs and cell lines simultaneously improved the prediction performance. In par-
ticular, combining chemical and structural drug properties, target information, and genomic
properties yielded more powerful drug response predictions than drug descriptors or targets
alone. Furthermore, KBMF achieved high accuracy in predicting missing drug responses,
allowing to construct a complete drug response map covering 116 drugs and 482 cell lines
from seven tissue types. The described method is able to tackle various relevant prediction
tasks related to drug response analysis, and other kinds of quantitative matrix prediction
from side information. It will help in further studies to suggest targeted experiments for
We are grateful to Suleiman A.Khan, Sohan Seth, Juuso Parkkinen, and John-Patrick Mpindi
for helpful comments. This work was financially supported by the Academy of Finland
(Finnish Center of Excellence in Computational Inference Research COIN, grant no 251170;
grant no 140057 and Biocenter Finland/DDCB). We acknowledge the computational re-
sources provided by Aalto Science-IT project and CSC-IT Center for Science Ltd.
Supporting Information Available
Benchmark experiment on QSAR data sets and their cross-validation results (Text, Tables
S-1 and S-2); Experimental details, evaluation criteria and additional results from integrative
and personalized QSAR analysis using the Sanger data set (Text, Tables S-3 to S-14, Figures
S-1 to S-5). This material is available free of charge via the Internet at http://pubs.acs.
(1) Garnett, M. J. et al. Systematic identification of genomic markers of drug sensitivity
in cancer cells. Nature 2012, 483, 570–575.
(2) Heiser, L. M. et al. Subtype and pathway specific responses to anticancer compounds
in breast cancer. Proc. Natl. Acad. Sci. USA 2012, 109, 2724–2729.
(3) Barretina, J. et al. The Cancer Cell Line Encyclopedia enables predictive modelling of
anticancer drug sensitivity. Nature 2012, 483, 603–607.
(4) Menden, M. P.; Iorio, F.; Garnett, M.; McDermott, U.; Benes, C.; Ballester, P. J.;
Saez-Rodriguez, J. Machine learning prediction of cancer cell sensitivity to drugs based
on genomic and chemical properties. PLoS One 2013, 8, e61318.
(5) Costello, J. et al. A community effort to assess and improve drug sensitivity prediction
algorithms. Nat. Biotechnol. 2014,
(6) Perkins, R.; Fang, H.; Tong, W.; Welsh, W. J. Quantitative structure-activity relation-
ship methods: Perspectives on drug discovery and toxicology. Environ. Toxicol. Chem.
2003, 22, 1666–1679.
(7) Wellcome Trust Sanger Institute, Genomics of Drug Sensitivity in Cancer. http://
www.cancerrxgene.org/, 2012; (accessed July 01, 2012).
(8) Myint, K. Z.; Xie, X.-Q. Recent Advances in Fragment-Based QSAR and Multi-
Dimensional QSAR Methods. Int. J. Mol. Sci. 2010, 11, 3846–3866.
(9) Shao, C.-Y.; Chen, S.-Z.; Su, B.-H.; Tseng, Y. J.; Esposito, E. X.; Hopfinger, A. J. De-
pendence of QSAR Models on the Selection of Trial Descriptor Sets: A Demonstration
Using Nanotoxicity Endpoints of Decorated Nanotubes. J. Chem. Inf. Model. 2013,
53, 142–158.
(10) Papa, E.; Villa, F.; Gramatica, P. Statistically Validated QSARs, Based on Theoret-
ical Descriptors, for Modeling Aquatic Toxicity of Organic Chemicals in Pimephales
promelas (Fathead Minnow). J. Chem. Inf. Model. 2005, 45, 1256–1266.
(11) Kraker, J. J.; Hawkins, D. M.; Basak, S. C.; Natarajan, R.; Mills, D. Quantitative
Structure-Activity Relationship (QSAR) modeling of juvenile hormone activity: Com-
parison of validation procedures. Chemom. Intell. Lab. Syst. 2007, 87, 33 – 42.
(12) Luilo, G. B.; Cabaniss, S. E. Quantitative Structure-Property Relationship for Predict-
ing Chlorine Demand by Organic Molecules. Environ. Sci. Technol. 2010, 44, 2503–
2508, PMID: 20230049.
(13) Matysiak, J. QSAR of Antiproliferative Activity of N-Substituted 2-Amino-5-(2,4-
dihydroxyphenyl)-1,3,4-thiadiazoles in Various Human Cancer Cells. QSAR Comb. Sci.
2008, 27, 607–617.
(14) Rogers, D.; Hopfinger, A. J. Application of Genetic Function Approximation to Quanti-
tative Structure-Activity Relationships and Quantitative Structure-Property Relation-
ships. J. Chem. Inf. Comput. Sci. 1994, 34, 854–866.
(15) Zheng, W.; Tropsha, A. Novel Variable Selection Quantitative Structure-Property Re-
lationship Approach Based on the k-Nearest-Neighbor Principle. J. Chem. Inf. Comput.
Sci. 2000, 40, 185–194, PMID: 10661566.
(16) Kompany-Zareh, M.; Omidikia, N. Jackknife-Based Selection of GramSchmidt Orthog-
onalized Descriptors in QSAR. J. Chem. Inf. Model. 2010, 50, 2055–2066.
(17) Cramer, R. D.; Patterson, D. E.; Bunce, J. D. Comparative molecular field analysis
(CoMFA). 1. Effect of shape on binding of steroids to carrier proteins. J. Am. Chem.
Soc. 1988, 110, 5959–5967.
(18) Hasegawa, K.; Funatsu, K. Evolution of PLS for Modeling SAR and omics Data. Mol.
Inf. 2012, 31, 766–775.
(19) Musumarra, G.; Condorelli, D. F.; Costa, A. S.; Fichera, M. A multivariate insight into
the in-vitro antitumour screen database of the National Cancer Institute: classification
of compounds, similarities among cell lines and the influence of molecular targets. J.
Comput.-Aided Mol. Des. 2001, 15, 219–234.
(20) Yamanishi, Y.; Pauwels, E.; Kotera, M. Drug Side-Effect Prediction Based on the
Integration of Chemical and Biological Spaces. J. Chem. Inf. Model. 2012, 52, 3284–
(21) Liu, P.; Long, W. Current Mathematical Methods Used in QSAR/QSPR Studies. Int.
J. Mol. Sci. 2009, 10, 1978–1998.
(22) Sutherland, J. J.; O'Brien, L. A.; Weaver, D. F. A comparison of methods for modeling
quantitative structure-activity relationships. J. Med. Chem. 2004, 47, 5541–5554.
(23) Lusci, A.; Pollastri, G.; Baldi, P. Deep architectures and deep learning in chemoin-
formatics: the prediction of aqueous solubility for drug-like molecules. J. Chem. Inf.
Model. 2013, 53, 1563–1575.
(24) Mullen, L. M.; Duchowicz, P. R.; Castro, E. A. QSAR treatment on a new class of
triphenylmethyl-containing compounds as potent anticancer agents. Chemom. Intell.
Lab. Syst. 2011, 107, 269 – 275.
(25) Lee, A. C.; Shedden, K.; Rosania, G. R.; Crippen, G. M. Data Mining the NCI60 to
Predict Generalized Cytotoxicity. J. Chem. Inf. Model. 2008, 48, 1379–1388, PMID:
(26) Gao, J.; Che, D.; Zheng, V.; Zhu, R.; Liu, Q. Integrated QSAR study for inhibitors
of hedgehog signal pathway against multiple cell lines:a collaborative filtering method.
BMC Bioinf. 2012, 13, 186.
onen, M. Predicting drug-target interactions from chemical and genomic kernels using
Bayesian matrix factorization. Bioinformatics 2012, 28, 2304–2310.
onen, M.; Khan, S.; Kaski, S. Kernelized Bayesian Matrix Factorization. In Proceed-
ings of the 30th International Conference on Machine Learning (ICML), Atlanta, USA,
16-21 June, 2013, 864–872.
olkopf, B.; Smola, A. J. Learning with Kernels: Support Vector Machines, Regular-
ization, Optimization, and Beyond ; MIT Press: Cambridge, MA, 2002; Chapter 15, pp
(30) Beal, M. J. Variational Algorithms for Approximate Bayesian Inference. Ph.D. thesis,
The Gatsby Computational Neuroscience Unit, University College London, 2003.
(31) Bolton, E. E.; Wang, Y.; Thiessen, P. A.; Bryant, S. H. PubChem: integrated platform
of small molecules and biological activities. Annu. Rep. Comput. Chem. 2008, 4, 217–
(32) Yap, C. W. PaDEL-descriptor: An open source software to calculate molecular descrip-
tors and fingerprints. J. Comput. Chem. 2011, 32, 1466–1474.
(33) National University of Singapore, PaDEL-descriptor: An open source software to cal-
culate molecular descriptors and fingerprints. http://padel.nus.edu.sg/software/
padeldescriptor/, 2011; (accessed January 15, 2013).
(34) Cruciani, G.; Crivori, P.; Carrupt, P.-A.; Testa, B. Molecular fields in quantitative
structure–permeation relationships: the VolSurf approach. J. Mol. Struct. 2000, 503,
(35) Pastor, M.; Cruciani, G.; McLay, I.; Pickett, S.; Clementi, S. GRid-INdependent de-
scriptors (GRIND): a novel class of alignment-independent three-dimensional molecular
descriptors. J. Med. Chem. 2000, 43, 3233–3243.
(36) Duran, A.; Martinez, G. C.; Pastor, M. Development and validation of AMANDA, a
new algorithm for selecting highly relevant regions in Molecular Interaction Fields. J.
Chem. Inf. Model. 2008, 48, 1813–1823.
an, A.; Zamora, I.; Pastor, M. Suitability of GRIND-based principal properties for
the description of molecular similarity and ligand-based virtual screening. J. Chem. Inf.
Model. 2009, 49, 2129–2138.
(38) National Institutes of Health, National Cancer Institute (NCI). http://www.cancer.
gov/, 1971; (accessed May 10, 2013).
(39) Hatzivassiliou, G. et al. RAF inhibitors prime wild-type RAF to activate the MAPK
pathway and enhance growth. Nature 2010, 464, 431–435.
Graphical TOC Entry
multiple cell lines
new cell line
data matrices
(biological, chemical & structural)
properties (GE, CNV, Mut)
data matrices
drugs target -prints 1&2D Vsurf GRIND2
responses
Integrative QSAR
Source: http://research.cs.aalto.fi/pml/online-papers/Ammad14jcim.pdf
Bangladesh J Microbiol, Volume 28, Number 1, June 2011, pp 7-11 Prevalence of Ciprofloxacin and Nalidixic Acid Resistant Salmonella entericaserovar Typhi in Bangladesh Shirin Afroj1, Mohammad Ilias1, Maksuda Islam2, and Samir K Saha2* 1Department of Microbiology, University of Dhaka, Dhaka-1000, Bangladesh. 2Department of Microbiology, Dhaka Shishu (Children) Hospital,Dhaka-1207, Bangladesh
ORAL APPLIANCES FOR THE TREATMENT OF SNORING AND OBSTRUCTIVE SLEEP APNEA: A REVIEW Oral Appliances for the Treatment of Snoring and ObstructiveSleep Apnea: A ReviewAn American Sleep Disorders Association Review Wolfgang Schmidt-Nowara1, Alan Lowe2, Laurel Wiegand3, Rosalind Cartwright4, Francisco Perez-Guerra5 and Stuart Menn6 1Pulmonary Division, Department of Medicine, University of New Mexico, Albuquerque, NM; 2Department of ClinicalDental Sciences, University of British Columbia, Vancouver, British Columbia, Canada; 3Department of Medicine,Pulmonary/Critical Care Division, Penn State College of Medicine, Hershey, PA; 4Sleep Disorders Service and ResearchCenter, Rush-Presbyterian-St. Luke's Medical Center, Rush University, Chicago, IL; 5Division of Pulmonary Disease, Scottand White Clinic, Temple, TX; and 6Division of Sleep Disorders, Scripps Clinic, La Jolla, CA







