Neuroglia in neurodegeneration

Neuroglia in neurodegeneration
Michael T. Henekaa,⁎, José J. Rodríguezb,e, Alexei Verkhratskyc,d,e,⁎
aKlinische Neurowissenschaften, Klinik und Poliklinik für Neurologie, Sigmund-Freud-Str. 25, 53127 Bonn, GermanybIKERBASQUE, Basque Foundation for Science, 48011, Bilbao, SpaincDepartment of Neurosciences, University of the Basque Country UPV/EHU, 48940, Leioa, SpaindFaculty of Life Sciences, The University of Manchester, Manchester, UKeInstitute of Experimental Medicine, ASCR, Prague, Czech Republic
Neuroglial cells are fundamental for control of brain homeostasis and they represent the
Accepted 19 November 2009
intrinsic brain defence system. All forms in neuropathology therefore inevitably involve
Available online 26 November 2009
glia. The neurodegenerative diseases disrupt connectivity within brain circuits affectingneuronal–neuronal, neuronal–glial and glial–glial contacts. In addition neurodegenerative
processes trigger universal and conserved glial reactions represented by astrogliosis and
microglial activation. The complex of recently acquired knowledge allows us to regard the
neurodegenerative diseases as primarily gliodegenerative processes, in which glial cells
determine the progression and outcome of neuropathological process.
2009 Elsevier B.V. All rights reserved.
Alzheimer's diseaseParkinson's diseaseDementiaAmyotrophic lateral sclerosis
Physiological functions of glial cells: Neuroglia as the brain homeostatic machinery . . . . . . . . .
2.1.1. Astrocytes organise the brain matter . . . . . . . . . . . . . . . . . .
2.1.2. Astrocytes form neuronal–glial–vascular units and provide neurones with metabolic support . .
2.1.3. Astrocytes control extracellular homeostasis of ions and neurotransmitters . . . . . . .
2.1.5. Astrocytes mould the CNS synapses and participate in synaptic transmission . . . . . . .
Microglia—the brain surveillance system. . . . . . . . . . . . . . . . . . .
Neuroglia determines the outcome of neurological pathology . . . . . . . . . . . . . . .
⁎ Corresponding authors. M.T. Heneka is to be contacted at Klinische Neurowissenschaften, Klinik und Poliklinik für Neurologie, Sigmund-
Freud-Str. 25, 53127 Bonn, Germany. A. Verkhratsky, The University of Manchester, Oxford Road, Manchester, M13 9PT, UK.
E-mail addresses: (M.T. Heneka), (A. Verkhratsky).
0165-0173/$ – see front matter 2009 Elsevier B.V. All rights reserved.
doi:
Neuroglia—the concept
features, demonstrate a profound functional heterogeneity indifferent brain regions and at different developmental stages.
The complexity of the cellular circuitry of human brain is
Nonetheless, each and every neuroglial cell has a conceptual
unparalleled by any other living system known so far. The
countenance to keep the brain homeostasis, starting from the
neural cells are exceedingly densely packed within a strictly
control of local molecular environment to providing the
limited volume of the skull, thus requiring a specific attention
intrinsic brain defence system. These glial homeostatic
to the control of brain homeostasis throughout early deve-
functions are many, and their failure inevitably signals brain
lopment and postnatal functioning. This specific requirement
pathology. This essay is specifically dedicated to the role of
is manifested in the highly developed brain–blood barrier,
neuroglia in neurodegenerative processes, yet prior to embark
which essentially limits the impact of bodily homeostatic
into the realms of disease we shall provide a brief overview of
systems on the central nervous system (CNS). The brain
physiological functions of main types of glial cells.
homeostasis therefore is entrusted to specific population ofneural cells known as neuroglia.
The concept of neuroglia as a connective tissue into which
Physiological functions of glial cells:
all elements of the central nervous system (CNS) are
Neuroglia as the brain homeostatic machinery
embedded was introduced by Rudolf Virchow ). In the course of
late 19th–early 20th century the cellular nature and morpho-functional heterogeneity of neuroglial cells were firmly
Astrocytes organise the brain matter
The astrocytes provide for the micro-architecture of the grey
matter by dividing it (through the process known as "tiling"
recent decades the functional relevance and versatility of
into relatively independent structural
neuroglia that is involved in all activities of the CNS, from
units. Each protoplasmic astrocyte dwelling in the grey matter
structural and metabolic support to information processing,
establishes its own territory within the limits of its elaborated
has started to be fully appreciated (
arbour of processes; this domain organisation exists in both
rodents and humans
these anatomical domains astroglial membranes cover syn-
the evolutionary uniqueness of human glial cells
aptic contacts and establish contacts with neuronal mem-
) indicates their role in the formation of human
branes as well as with blood vessels. Evolution of the CNS and
appearance of the intellect coincided with a remarkable
The neuroglia appeared early in evolution, when primitive
increase in the size and complexity of astroglial cells
nervous systems began to emerge, and became the predomi-
That is the average diameter of the
nant cellular type in the brain of Homo sapiens. The main types
domain belonging to a human protoplasmic astrocyte is ∼2.5
of neuroglia are represented by astrocytes (named so by
times larger that the domain formed by a rat astrocyte (142 vs.
Michael von Lenhossek )), oligodendrocytes
56 μm). The volume of the human protoplasmic astrocyte
(christened by Pio del Rio-Hortega NG2
domain was ∼16.5 times larger than that of the corresponding
positive glia (initially revealed by William Stallcup (
domain in a rat brain. Furthermore, human protoplasmic
) and identified as a separate class of
astrocytes have ∼10 times more primary processes emanating
glia in the recent decade (
from their somatas, and correspondingly much more complex
and microglia (discovered by Rio-
processes arborisation. Likewise, the fibrous astrocytes, pop-
Hortega The cellular elements
ulating the white matter are ∼2.2 times larger in humans
comprising each of these groups, although having common
when compared to rodents. The astroglial domains that
parcellate the grey matter can be the unifying structures,
port into the astrocytes. An increase of Na+ concentration in
which, by covering many synapses (about 20.000–120.000 in
the cytosol of astrocytes stimulates glycolysis, which results
rodents and 270.000 to 2.000.000 in humans), can integrate and
in synthesis of lactate; the latter is then transported to
regulate the activity of large synaptic sets (
neurones by monocarboxylase transporters 1 and 2, thus
providing active cells with much needed energy substrate
The single astrocytic domains are further integrated by
virtue of gap junctions ) into astroglial syncytia. The gap junctional
Astrocytes control extracellular homeostasis of ions and
contacts are localised on the peripheral processes where two
astroglial domains overlap; the actual areas of contacts
Control of the extracellular concentrations of ions, metabo-
between neighbouring astrocytes are quite small. It is
lites and neuroactive molecules is of a paramount importance
interesting that the overlapping areas of human astrocytes
for brain function. Neuronal excitability is maintained by
were ∼20 times larger than in rodents
relatively large transmembrane fluxes of ions, which are
which possibly indicates a higher degree of coupling. Gap
moved by electro-chemical gradients. These fluxes affect the
junctions provide for a glial information-transfer system, as
extracellular concentrations of ions, which in turn change
they form pathways for intercellular diffusion of many
these gradients. The accumulation of extracellular K+ is
molecules, which convey the long-range signalling. One of
particularly important as it accompanies the repolarisation
these signalling pathways, represented by diffusion of second
phase of action potentials; under physiological conditions the
messenger InsP3 with subsequent Ca2+ release is well char-
extracellular potassium concentration ([K+]o) can rise up to 10–
acterised as propagating Ca2+ wave
12 mM (Control of extracellular K+
both Ca2+ release
concentration is accomplished by astrocytes through local K+
and Ca2+ waves are considered to be a substrate for astroglial
uptake involving inward rectifier K+ channels and K+ spatial
The spatial K+ buffering provides for the
). However, other molecules, such as,
redistribution of K+ from the areas with elevated [K+]o to the
for example, metabolic substrates ) can
regions with low [K+]o. This spatial buffering occurs either in
form alternative inter-glial signalling systems. The astroglial
glial syncytia or within the confines of single radial Muller glial
syncytia are formed within larger anatomical structures: for
cells, through the process known as K+ siphoning (
example, in the somatosensory cortex, astroglial networks
are confined to individual barrels with very weak (if any)
The astroglial transport of ions, which accompanies
inter-barrel coupling (
neuronal activity (e.g. K+ uptake through KIR channels, or Na+
accumulation alongside with glutamate transportation)requires concomitant movement of water. Control of water
Astrocytes form neuronal–glial–vascular units and
homeostasis is also accomplished by astrocytes. High synaptic
provide neurones with metabolic support
activity is associated with local shrinkage of the extracellular
The concept of astrocytes forming a metabolic connection
space, which is regulated by water transport across astroglial
between neurones and blood vessels was introduced by
membranes and water redistribution through the glial syncy-
Camillo Golgi after his discovery of astroglial endfeet embrac-
tium. Water enters and leaves the astroglial syncytia through
ing brain capillaries (). The astroglial domains are
aquaporins channels (mostly of AQP4 type) which are
instrumental in establishing such a connection through
concentrated in perisynaptic processes and in the perivascular
moulding the neuronal–glial–vascular units, which integrate
and subpial endfeet Further-
neural circuitry with local blood flow. Indeed, most protoplas-
more, K+ buffering and water redistribution are tightly
mic astrocytes contact neighbouring capillaries through the
coordinated and alteration of water flux impairs upon K+
perivascular processes forming an endfoot (
buffering ().
). Astrocytes also forge a functional link between neu-
Astrocytes play the central role in the extracellular
rones and blood vessels. An increase in focal neural activity
homeostasis of neurotransmitters and most importantly of
triggers rapid vasodilatation, the phenomenon known as
glutamate. Glutamate, despite being the main excitatory
functional hyperaemia An increase
transmitter in the CNS, is the most powerful neurotoxin, and
in neuronal activity within the astroglial domain triggers Ca2+
every excess of glutamate in the extracellular spaces triggers
signals, which enter astrocyte endfeet and result in the release
excitotoxic neuronal death. Astrocytes are the main sink of
of vasoactive substances. The latter can trigger either vasocon-
glutamate in the brain; from the bulk of glutamate released
striction or vasodilatation
during synaptic transmission, about 20% is accumulated into
postsynaptic neurones and the remaining 80% is taken up by
perisynaptic astrocytes
The metabolic support of neurones is achieved through a
glucose-lactate shuttle operative within the astroglial
Glutamate transport is accomplished by specific glutamate
domains. Astrocytes accumulate about 50% of glucose enter-
transporters (represented by
ing the brain tissue, and store it in the form of glycogen. An
several types (EAAT1 to EAAT5 in human brain). The EAAT1
increase in neuronal activity, accompanied with an increased
and EAAT2 (known in rodent brain as glutamate/aspartate
glutamate release, results in Na+-dependent glutamate trans-
transporter, GLAST, and glutamate transporter-1, GLT-1) are
expressed exclusively in astrocytes and are
which are absent in hippocampus
responsible for the bulk of glutamate uptake. The transloca-
). Gliotransmitter
tion of glutamate is powered by transmembrane ion gradients,
release actively modulates synaptic transmission by activat-
and the transport of a single glutamate molecule requires an
ing various neuronal receptors such as, for example, NMDA or
influx of three Na+ ions and one H+ ion coupled with the efflux
adenosine receptors (
As a result Na+/glutamate transporter is electrogenic and its
Astrocytes do not only participate in synaptic transmission—
activation produces a net inward sodium current
they act as key elements in synaptogenesis, in synaptic
), which may substantially affect intracellular Na+
maturation and maintenance. In the in vitro condition, the
concentration. The excessive sodium accumulation accompa-
addition of astrocytes triggers very substantial increase (up
nying glutamate accumulation can be counterbalanced by Na+
to seven times) in synapse formation
efflux through Na+/Ca2+ exchanger working in the reverse
Astrocytes produce and secrete cholesterol
which is critically important for synapse formation
Astrocytes are also crucial for the recovery of glutamate to
and secrete variety of factors needed for both synaptic
the presynaptic terminal. After entering the astroglial cells
maturation and maintenance
glutamate is converted into glutamine by the astrocytic-
). Furthermore astrocytes synthesise and release
specific glutamine synthetase
thrombospondins 1 and 2 that promote synaptogenesis both
). Glutamine, being non-toxic, can subsequently be safely
in vivo and in vitro and are critically important for post-lesion
transported back to the presynaptic terminal through the
synaptic plasticity, remodelling and regeneration (
extracellular space; after entering the neuronal compartment
glutamine is converted into glutamate, thus accomplishingthe glutamate–glutamine shuttle.
The function of oligodendrocytes is to produce the myelin
The ability of astrocytes to release chemical transmitters
sheaths that insulate axons in the CNS. The myelin sheath is
(named gliotransmitters in order to distinguish them from
composed from several specific proteins including myelin
neurotransmitters) is fundamental for their involvement in
proteolipid protein (PLP), myelin basic protein (MBP) and
information processing in neuronal–glial networks. The
myelin associated glycoprotein (MAG)
gliotransmitters include glutamate, ATP, D-serine, GABA,
. The myelin sheath being a fatty
taurine and possibly other molecules (see
insulating layer facilitates the saltatory conduction of action
potentials (So far four different phenotypes (I–IV)
subdivide oligodendrocytes. Developmentally, all four types
for review). The gliotransmitters
of oligodendrocytes are likely to originate from common
can be released from astrocytes through Ca2+-dependent
oligodendrocyte progenitor cells (OPCs) nestled in the SVZ.
exocytosis (), by diffusion though large pore
After the migration to their target regions OPCs differentiate
channels (e.g. P2X7 receptors, hemichannels or volume-
and mature up-regulating the expression of myelin proteins,
activated Cl− channels
and begin to form the myelin sheath
transporters (reversed glutamate transporter () or a cystine-glutamate antiporter
Astrocytes mould the CNS synapses and participate in
The NG2 glial cells (identifiable by the expression of NG2
synaptic transmission
chondroitin sulphate proteoglycan (which
Most of the CNS synapses are formed by three elements—the
are also known as synantocytes (from the Greek synanto for
astroglial perisynaptic process, the presynaptic neuronal
contact ()), are present throughout the
terminal and the postsynaptic neuronal membrane—the
developing and adult brain (
structure generally known as a tripartite synapse (
). Although NG2 cells express various markers,
which are characteristic for oligodendrocyte progenitor cells,
has a dual role in this tripartite synapse. First, by the virtue of
they have some distinct features, which permit classifying
neurotransmitter receptors expressed in the astroglial mem-
them as a separate type of glia. The NG2 cells have a stellate
brane, the astrocyte can sense the transmitter release from
morphology with many primary processes, which bifurcate to
the neuronal terminal, and secondly by releasing gliotrans-
form a process arborisation with a diameter of about 100 μm
mitters the astrocyte can modulate the efficacy of the synapse.
Astroglial cells can potentially express virtually every neuro-
Physiologically many NG2 cells express voltage-gated Na+
transmitter receptor (
channels which, at
least in cortical NG2 cells are dense enough to generate action
). This expression however, is strictly controlled in vivo
potentials (In addition, the NG2 glia
and astrocytes from different brain regions are endowed with
express Ca2+ permeable AMPA receptors, GABA receptors (
very distinct complement of receptors. The cortical astrocytes,
and, most likely, purinoceptors
for example, express functional NMDA and P2X1/5 receptors,
In the hippocampus the NG2 glia receive functional
synaptic inputs from CA3 pyramidal neurones and GABAergicinterneurones (). The NG2 cells may be
Neuroglia determines the outcome of
important for integration in the brain because their processes
neurological pathology
pass through several neuronal layers and traverse grey andwhite matter. Finally, the NG2 glial cells are highly plastic
Glial cells are fundamental for the control of brain homeosta-
progenitor cells that can give rise to astrocytes
sis, and they represent the intrinsic brain defence system.
) and may be even to neurones.
First, the homeostatic systems expressed in astrocytesprevent homeostatic imbalances triggered by various types
Microglia—the brain surveillance system
of stressors applied to CNS. Second, two types of glia—theastrocytes and microglia—possess evolutionary conserved
Microglial cells are the resident macrophages of the CNS.
programs of activation in response to brain damage. A variety
Microglia constitute around 10% of all cells in the nervous
of brain insults trigger a condition generally referred to as
system. These cells are of myeloid origin (
reactive gliosis, which includes astrogliosis and activation of
) and they enter CNS during the early postnatal period
microglia. The astrogliosis (
through the so-called "fountains of microglia" (
is essential for both limiting the areas of
). After entering the CNS, these cells disseminate through
damage (by scar formation through anisomorphic astrogliosis)
the parenchyma and transform into the resting microglia. The
and for the post-insult remodelling and recovery of neural
resting microglial cells have small somatas and multiple fine
function (by isomorphic astrogliosis). The activation of micro-
processes. Every microglial cell occupies the defined territorial
glia is fundamental for the brain immune response as well as
domain, which does not overlap with neighbouring microglia.
for the removal of both invading infectious agents and
In the physiological conditions, microglial processes are
posthumous cell debris (
constantly moving scanning the microenvironment in their
anatomical domains (
All these glial defence mechanisms are genuinely surviva-
). Microglia represent the innate immune system in the
listic, and yet, the glial cells being homeostatic tools possess
brain and thus are the first line of defence against invading
(as many other biological homeostatic systems do) an
pathogens and serve as specialised sensors for brain tissue
inherent dichotomy—they can be protective as well as
deleterious In fact, stronger brain insults may push
). Insults to the nervous system trigger a complex and
glial homeostatic systems towards a damage exacerbating
multi-stage activation of microglia, which results in both
mode. The severe stress on astroglial energetics with a
phenotypic and functional changes (
subsequent loss of ion homeostasis may trigger a massive
). This process manifested by
release of glutamate (through reversed transporters or large
microglia transition from a surveillance state to an activated
pore channels), a substantial leak of K+ ions, release of NO and
state is controlled by multiple extracellular signals acting
reactive oxygen species—i.e. agents promoting neurotoxicity
through a multitude of receptors. These "danger" signals are
The activation and over-
represented either by the disappearance of certain molecules,
activation of microglia may have similar deleterious effects
indicative of normal brain functioning or by the appearance of
through both phagocytic activity and release of pro-inflam-
new molecules associated with infectious agents, debris from
matory and neurotoxic factors. All these glial reactions are
damaged or dying cells or misfolded and aggregated proteins
intimately involved into acute brain damage such as trauma
appearing in response to a primary degenerative process
or stroke. All in all "glia appears as a brain warden, and as such
(the concept of "on" and "off" signalling—
it is intrinsically endowed with two opposite features: it
protects the nervous tissue as long as it can, but it also can act
Under pathological situations, such as neurodegenerative
as a natural killer, trying to eliminate and seal the damaged
diseases, strokes, traumatic injuries and tumour invasions,
area, to save the whole at the expense of the part (
these cells become activated, migrate to and surround
damaged or dead cells, and subsequently clear cellular debris
Neuroglia is also thoroughly involved in pathogenesis of
from the area, similarly to the phagocytic macrophages of the
many chronic neurological disorders
peripheral immune system ().
Astrocytes in epileptic foci in the
Activated microglia up-regulate a variety of surface receptors,
temporal lobe epilepsy undergo both morphological and
including major histocompatibility complex and complement
functional changes (see
for review). In humans, epilepsy triggers reactive astrogliosis
). They also undergo fundamental morphological changes
and an increase in GFAP expression
from a ramified phenotype to motile activated amoeboid cells
). The role of glutamate release from
(Once they are immunostimulated in
astrocytes in synchronous discharges triggering epileptiform
response to neurodegenerative events, these microglia cells
seizures has been proposed (although the
release a variety of proinflammatory mediators including
exact degree of astroglial involvement in this process remains
cytokines, reactive oxygen species, complement factors,
controversial (). At any rate astrocytes from a
neurotoxic secretory products, free radical species and NO,
human epileptic brain display spontaneous Ca2+ oscillations
all of which can contribute to neuronal dysfunction and cell
and have an increased gap
death, ultimately creating a vicious cycle (
junctional coupling which indicates remodel-
ling of signalling cascades. In addition, astroglial glutamate
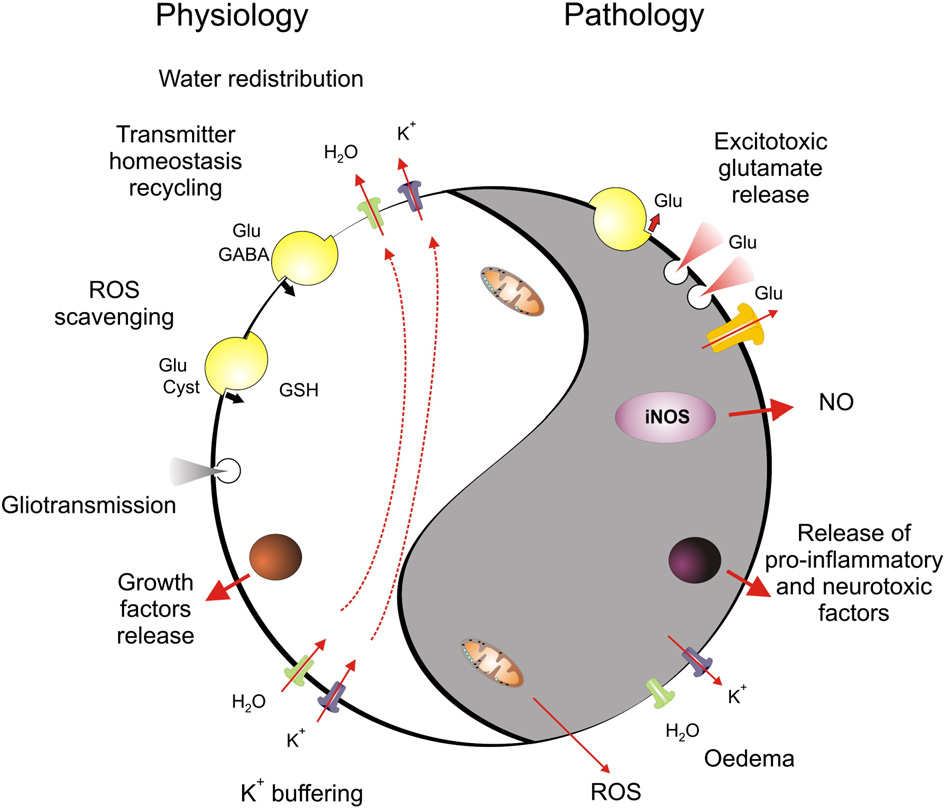
Fig. 1 – Dual role of astroglial homeostatic cascades. The homeostatic cascades expressed in astrocytes control extracellular ionhomeostasis through K+ buffering, regulate movements and distribution of water, control extracellular concentration ofneurotransmitters and provide main reactive-oxygen species scavenging system. In pathological conditions, when astrocytesexperience metabolic stress, the same systems may contribute to brain damage. Failure in water transport triggers brainoedema, reversal of neurotransmitter transporters together with Ca2+-dependent exocytosis and opening of high-permeabilityplasmalemmal channels contributes to glutamate excitotoxicity; inadequate K+ buffering promotes further overexcitation ofneural cells, and glial cells begin to release ROS and pro-inflammatory factors, further exacerbating brain damage.
transport can also be important for controlling seizures
their degeneration and profound damage of the white matter
development, as was directly demonstrated in genetically
that may result in severe dementia. A particular type of post-
modified mice, which lack astroglial transporter GLT-1. These
stroke dementia is represented by Binswanger's disease (or
animals develop spontaneous and lethal seizures which killed
subcortical dementia), which is a form of vascular dementia
half of homozygous mice before they reach 6 weeks of age
characterised by diffuse white matter lesions; it leads to
progressive loss of memory, cognition and behavioural adapta-
Astroglial cells are also involved in a variety of psychiatric
tion (). The infarct occurring in
disorders. The loss of astrocytes was observed in patients
white matter triggers progressive death of oligodendrocytes,
suffering from depression The
activation of astrocytes and microglia and degeneration of axons
astrocytes may also play a role in the pathogenesis of
). The primary pathological steps most likely
schizophrenia, through their control over glutamate homeo-
are associated with ischaemic death of oligodendrocytes.
stasis and gliotransmission. The recently developed glutamate
Another ischaemia-related disease arising from death of
theory of schizophrenia ) stresses the role
oligodendrocytes is periventricular leucomalacia; a condition
of the hypofunction of NMDA receptors, which are under
that causes diffused cerebral white matter injury
positive control of the gliotransmitter D-serine. The latter was
This occurs mostly in prematurely born infants. The
used in clinical trials with certain beneficial effects
roots of this pathology can be found in (1) poor vascularisation
), once more indicating that deficient gliotransmission
of white matter in premature infants and (2) the prevalence of
may be involved in schizophrenia pathogenesis.
oligodendrocyte progenitors, which are particularly sensitive
Pathological changes in oligodendroglia are central to the
to ischaemia, reactive oxygen species and glutamate excito-
broad class of diseases of white matter. Oligodendrocytes and
toxicity. Thus, periods of even comparatively mild ischaemia
oligodendrocyte precursors are highly vulnerable to excitotoxic
result in profound damage to white matter and the demise of
insults. This excitotoxic death is mediated by Ca2+ influx
many oligodendrocyte progenitors. This, in turn, leads to
following overactivation of ionotropic glutamate receptors and
defective myelination, which further alters cerebral cortex
possibly P2X7 purinoceptors
development and leads to the impairment of pyramidal tracts,
The oligodendroglial death can directly affect axons, causing
with subsequent neurological disorders, including cerebral
palsy and cognitive deficits. Last but not least, the oligoden-
hypothesis of direct toxicity of the mutant gene on neurones
droglia plays a central role in various demyelinating disorders,
mediated through reduced free-radical buffering was not
including multiple sclerosis. The pathogenesis and molecular
confirmed while recent studies
mechanism of these diseases was a subject of many reviews
have demonstrated the key role of glial impairments in the
ALS pathogenesis.
The ALS is associated with astrogliosis and microglial
) to which we address the curious reader.
activation, which was described in both humans and transgenic
Finally, the microglia controls the immune response and
phagocytotic after-damage clearance system of the brain.
The astrogliosis, however, is preceded by astroglial degenera-
Through multiple stages of activation, microglia grades the
tion and atrophy, which occurs before neuronal death and the
reaction to brain lesion, being one of the most important
appearance of clinical symptoms in the hSOD1G93A transgenic
determinants of the course of CNS pathology
mouse At later stages of the disease the
reactive astroglia appears, although atrophic astrocytes are also
Our knowledge about the pathological potential of neuro-
present close to lesion sites. The astroglial degeneration was a
glia is still rudimentary, as major attention has always been
property of SOD1 bearing astrocytes, which demonstrated an
diverted to the pathology of neurones. Nonetheless, it becomes
increased vulnerability to glutamate, mediated through
increasingly obvious that it is the glia which determine the
mGluR5 receptors ). The hSOD1 bearing
initiation, course and outcome of majority (if not all) diseases
astrocytes also release neurotoxic factors and assist microglial
of the nervous system. Indeed, loss of glial support inevitably
signals neuronal demise, and glial performance decides upon
). Finally, selective silencing of the SOD1 mutant
the balance of neuroprotection, neuroregeneration and neural
gene in astrocytes significantly slowed the progression of ALS
death, thus controlling the pathology of the brain. Specifically,
in transgenic mice (). Notably, a recent
glial reactions are instrumental in shaping various neurode-
PET study by targeting MAO-B, which in
generative processes, which we shall discuss in subsequent
the CNS is almost confined to astrocytes, using C11 (L)-deprenyl,
chapters that are specifically dedicated to the role of astroglia
described a profound astrocytic proliferation in ALS patients.
and microglia in neurodegenerative processes.
Therefore, astrocytes can be considered as central players
in APS pathology. At the initial stages, glutamate inducesgliotoxicity. The atrophic astrocytes in turn reduce synaptic
Astrocytes in neurodegeneration and AD
coverage and fail to perform their homeostatic and neurone-supportive functions. This initiates neurodegeneration, which
The causes of neurodegenerative diseases are many, from
triggers reactive gliosis; reactive astrocytes release neurotoxic
traumatic or infectious attacks to intrinsic processes associ-
factors and stimulate microglial activation thus supporting
ated with genetic predispositions or the accumulation of
the vicious circle of neurodegeneration.
sporadic errors of yet unknown origins. The neurodegenera-tive disorders, which affect the main human asset, the
Wernicke encephalopathy
intellect, are in essence the failures of connectivity withinbrain circuitry. The astrocytes, being involved in synaptic
The combination of ataxia, ophthalmoplegia, and mental
birth, maturation and maintenance, as well as in controlling
changes, reflecting encephalopathy with deep thalamo-corti-
the brain homeostasis and neurotransmitters balance, are
cal lesions was initially described by Carl Wernicke
strategically important for preserving connection in brain
This encephalopathy is generally caused by a
networks, and their malfunction can be critical for the
deficiency of thiamine. Although the nature of neuronal death
development of neurodegeneration. Indeed, recently the
remains unclear, the possible failure of glutamate homeosta-
pathological potential of astroglia in neurodegenerative dis-
sis can assume the leading role. Indeed, specific analysis
eases started to be experimentally revealed.
revealed a substantial reduction (60–70%) of the expression ofastroglial transporters EAAT1 and EAAT2 in cortical samples
Amyotrophic lateral sclerosis
from human tissues obtained from confirmed cases ofWernicke encephalopathy. A similar profound decrease in
Amyotrophic lateral sclerosis (ALS, known in the United States
astroglial glutamate transporters was found in the rat
as "Lou Gehrig's disease"; named so after a baseball player
thiamine deficiency model of the disease ().
who suffered and died from this pathology) was described by
The failure of astroglial glutamate uptake can be the reason for
Jean-Martin Charcot in 1869 (
neuronal excitotoxicity and subsequent lesions. In addition, a
). The ALS is manifested by degeneration of
significant decrease in expression of GFAP, astrocytic gluta-
motor neurones located in the cortex, in the brain stem and
mine synthetase and astrocytic GAT-3 GABA transporter, all
in the spinal cord. Clinically the ALS appears in the form of
indicative of astroglial dystrophy or death, was observed in the
progressive paralysis and muscle atrophy. The ALS appears in
thalamus of thiamine deficient rats ().
both familial (∼10% cases) and sporadic forms. About 20% ofALS cases are associated with dominant mutations in the gene
Parkinson's disease
coding for Cu–Zn superoxide dismutase (SOD1) ); this mutated gene become instrumental in generating
The disturbed locomotive and motor functions (which include
animal models of ALS (The initial
akinesia, rigidity, tremor at rest, and postural abnormalities) are
prevailing clinical symptoms of the Parkinson's disease (PD—
astrocytes is observed ); the degree of glial
)). These symptoms arise from
atrophy displayed direct correlation with the severity of
the specific extermination of dopaminergic neurones in sub-
dementia. In another study however, prominent astrogliosis
stantia nigra with a subsequent severe impairment of nigros-
and profound increase in astrocyte density (up to four to five
triatal dopaminergic transmission. The systematic investigation
times) was found in post-mortem tissues
of the astroglial involvement into the pathogenesis of PD has not
yet been performed, although existing data allow suspecting the
Early and prominent astrogliosis also accompanies tha-
pathological potential for astrocytes. At the late stages of the
lamic dementia. In this form of pathology a specific prolifer-
disease, profound astrogliotic changes were identified in sub-
ation of perivascular and perineuronal astroglial processes
stantia nigra, reflecting the inflammatory state accompanying
changes are observed. These changes in astroglia are consid-
neurodegeneration
ered to be the primary pathological change, which can
). The early changes in astroglia are unknown
produce dementia even in the absence of severe neuronal
and yet they may play an important role in the progression of PD.
The substantia nigra has less astrocytes compared to other
Astrocytes (and microglia) also play a primary neurotoxic
brain region; therefore it is tempting to speculate that when
role in immunodeficiency virus-1 (HIV-1) associated dementia,
stressed these astrocytes fail and cease to support the dopami-
or HAD In HAD significant astrogliosis and an
nergic neurones, which in turn contributes to the degeneration
increase in GFAP expression is observed in the entorhinal cortex
of the latter. The support and protection of dopaminergic
and the hippocampus The progression of
neurones by astroglia is well documented in vitro
HAD, however, leads to a significant astroglial cell loss through
). The addition of
apoptosis, which is specifically prominent in the subjects with
astrocytes to midbrain cultures increases the percentage of
rapidly progressing cognitive deficits In
tyrosine hydroxylase-positive (i.e. dopaminergic neurones)
this type of pathology, the virus infects exclusively microglia,
from 2–5% to ∼40%; the effect, which is mimicked by glial-
although the dementia progresses due to NMDA-receptor
conditioned medium collected from cultures of mesencephalic
mediated neuronal death through necrosis or apoptosis (
astrocytes The same medium also protects
). The glutamate excitotoxicty can result from TNF-α
dopaminergic neurones against cell death triggered by 1-
release from infected and activated microglia. This triggers a
methyl-4-phenylpyridinium (MPP+; the latter is a selectively
massive release of glutamate from astrocytes following a TNF-
toxic for dopaminergic neurones and is the active agent in 1-
α-mediated activation of chemokine receptors of the CXCR4
methyl-4-phenyl-1,2,3,6-tetrahydropyridine, MPTP induced aki-
type expressed in astroglial membranes ).
netic rigid syndrome, considered to be the relevant animal
Incidentally, the same CXCR4 receptors can also be activated by
model of PD) or by NO (promotes neurite
the isoform of HIV-1 coat protein gp120IIIB, implicated in HAD
growth and affects signalling cascades in these neurones (
pathology. The neurotoxicity can also be exacerbated by the
release of additional inflammatory and death factors from both
Furthermore, astrocytes play a central role for L-DOPA-
astrocytes and activated microglia
dependent PD therapy. The L-DOPA (the main agent used in
clinical treatment of PD) is toxic for dopaminergic neurones in
Various types of non-AD dementia (e.g. progressive supra-
nuclear palsy, corticobasal degeneration and Pick's disease)
), albeit it is beneficial for these neurones in vivo.
are associated with the appearance of tau protein inclusions in
The difference is all in astroglia, and indeed addition of glial-
astroglial cells, which normally express very little (if at all) of
conditioned medium to cultures of dopaminergic neurones
tau protein ). A targeted expression of FTDP-17
prevented L-DOPA neurotoxicity and turned it into trophic
tau protein (the FTDP-17 gene is associated with parkinsonism
and frontotemporal dementia linked to chromosome 17) into
Finally, astrocytes may promote differentia-
astrocytes in a transgenic mouse model triggered age-depen-
tion of stem cells into dopaminergic neurones and facilitate
dent neurodegeneration, thus directly indicating that astroglia
their incorporation into the neuronal circuitry
can indeed be a primary cause of a chronic neurodegenerative
Therefore, the early astroglial atrophy and failure to
support dopaminergic neurones may be an important patho-
Alzheimer's disease
logical step in the development of PD.
The glial involvement in the pathogenesis of Alzheimer's
disease (AD) was initially suggested by Alois Alzheimerhimself (He had demonstrated that the
Astrocytes are affected in many types of dementia. Depending
neuritic plaques (the extracellular deposits of fibrillar β-
on the type and progression of the disease, both astroglial
amyloid, which together with the tau neurofibrillary tangles
atrophy and astrogliosis are observed; these two processes can
represent the major histopathological markers of AD) include
develop in parallel depending on the pathological stage. In the
glial cells (). The AD brains are characterised by
frontotemporal dementia (the clinical term covering several
prominent astrogliosis, mostly observed in the cells surround-
types of sporadic non-Alzheimer cognitive disruptions, which
ing amyloid plaques with processes of activated astrocytes
include e.g. Pick's disease and frontotemporal lobar degener-
participating in formation of neuritic plaques
ation) early and dramatic apoptotic death and dystrophy of
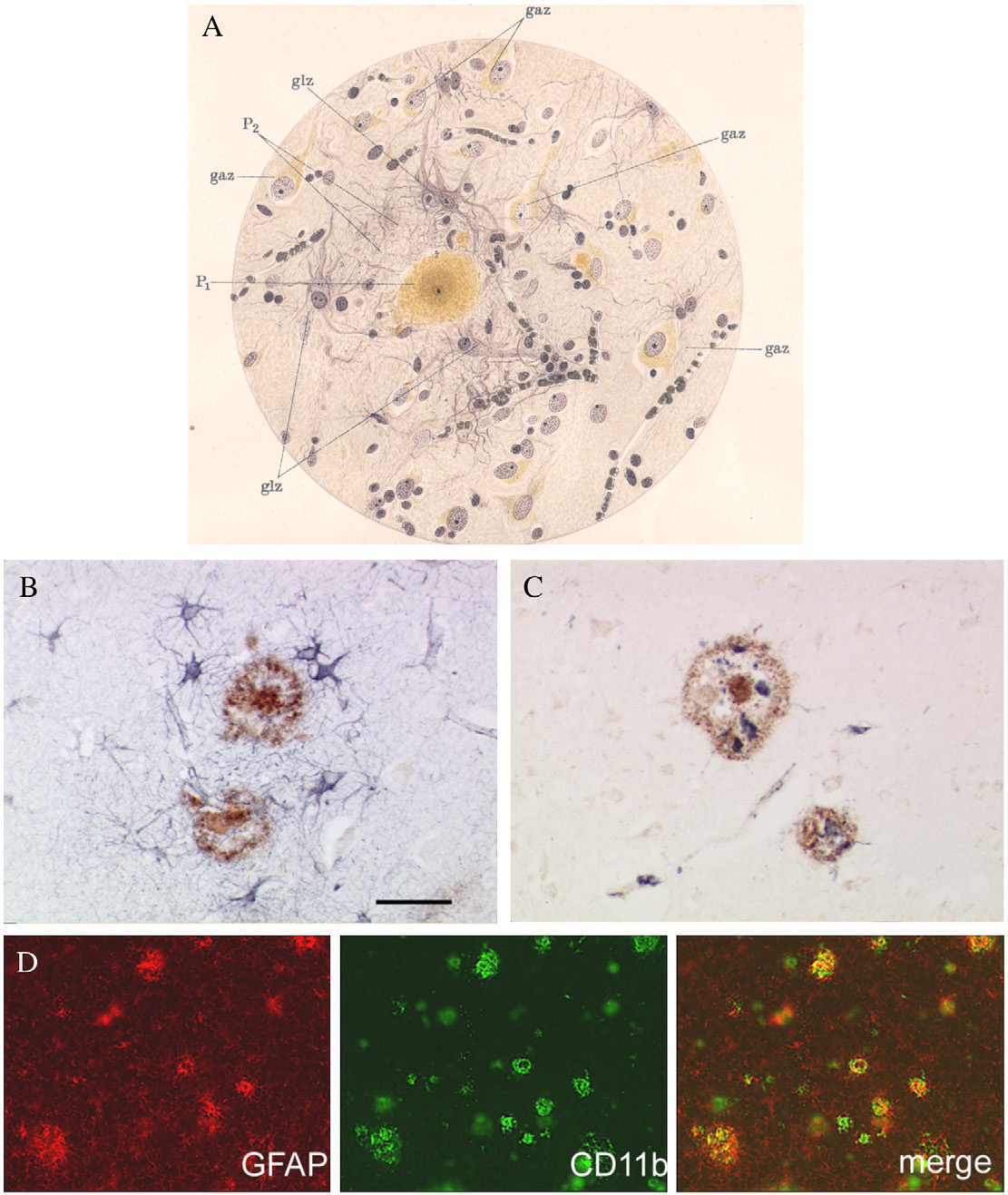
Fig. 2 – Activation of glial cells at sites of β-amyloid deposition in human brain and in APP transgenic mice. (A) Neuritic plaquesas seen and drawn by Alois Alzheimer (The plaque is surrounded by activated astrocytes; and activatedmicroglia is present at the peripheral region. Abbreviations: P1—the central part of the plaque (amyloid deposition);P2—periphery of the plaque; gaz—neurone; glz—glial cell. (B) Double immunostaining of a human brain section derived from a70 years old Alzheimer disease (AD) patient reveals GFAP positive astrocytes (blue) surrounding 6E10 positive β-amyloiddeposits (brown). (C) Double-staining for β-amyloid (brown) and CD68-positive microglia cells. Scale bar = 50 μm. (D) Doubleimmunostaining of GFAP and CD11b in a cortical section of a 12 month old APP23 transgenic mouse shows a focal and closeassociation of both markers for astro- and microglial reactivity.
The β-amyloid peptide presents activating signals for
The Aβ-induced [Ca2+]i oscillations lasted for many hours and
astrocytes; the exposure of cultured glial cells to aggregated
were linked to neuronal death, which occurred 24 h after the
β-amyloid or to amyloid plaques isolated from human AD
administration of Aβ to the cultures. The inhibition of [Ca2+]i
brains trigger reactive astrogliosis The Aβ
oscillations prevented neuronal death ().
also induces functional changes in astrocytes in vitro: the β-
In the same mixed culture model Aβ was also shown to induce
amyloid peptide (Aβ1–42) and its toxic fragment (Aβ25–35)
mitochondrial depolarisation and oxidative stress in astro-
induced spontaneous [Ca2+]i elevations and [Ca2+]i oscillations
cytes; the release of reactive oxygen species from stressed
in astrocytes growing in mixed astroglial–neuronal cultures.
astrocytes caused neuronal death ().
The abnormalities in astroglial Ca2+ signalling were ob-
Nevertheless, the astrogliosis is not the only astroglial
served in the brains of transgenic AD mice. In these experi-
reaction in the AD brains. In our recent studies, performed on
ments, employing in vivo multiphoton confocal microscopy,
different regions of the brains of triple-transgenic (3x-Tg-AD—
the general elevation of resting [Ca2+]i was observed through-
mice, both astrogliosis and astroglial
out the astroglial syncytia. In addition, astrocytes located in
atrophy were found (; Rodriguez and
the vicinity of plaques triggered spontaneous long-distance
Verkhratsky, papers in preparation; The decrease in
propagating Ca2+ waves, which were absent in control animals
complexity of astrocytes, which indicated their atrophy, began
to be observed before the formation and consolidation of
The participation of astrocytes in plaque formation initi-
neuritic plaques. In a plaque infested brain the reactive
ated the hypothesis of the Aβ-clearing role of astroglia (
astrocytes were concentrated around the Aβ plaques, whereas
); with subsequent astroglial degeneration
astroglial cells distant to the plaques had an atrophic features.
triggered by the accumulated β-amyloid peptide. Indeed, theplating of isolated healthy astrocytes on the slices preparedfrom transgenic (APP) AD mice resulted in astrocytes migra-
Microglia in neurodegeneration and AD
tion towards the plaques with subsequent accumulation anddegradation of Aβ. To support this finding, some evidence
Amyotrophic lateral sclerosis
suggests that astroglial cells are able to phagocyte Aβpeptides, a process which may depend on their apolipoprotein
The primary pathological feature of ALS is the loss of motor
E (ApoE) status, suggesting that ApoE polymorphisms may
neurones (which is accompanied by a robust
influence the risk to develop AD by affecting astroglial Aβ
glial response including the activation of microglia and
phagocytosis ). In contrast,
astrocytes as well as the expression of cyclooxygenase 2
endogenous astrocytes surrounding the Aβ plaques were
(COX-2) and nitric oxide synthase (iNOS) in the spinal cord
unable to accumulate and remove Aβ (
). In the triple transgenic mouse model of AD (3xTg-AD;
). The histological studies of post-mortem
harbouring the mutant genes for amyloid precursor protein
brains and spinal cord tissue were recently supported by a
(APPSwe), presenilin 1PS1M146V and tauP301L ())
study using the PET ligand PK1195, which labels the peripheral
very little (if any) Aβ accumulation by reactive astrocytes was
benzodiazepine receptor being expressed by activated micro-
observed (These data clearly indicate
glia in the brain. In this study, found
the phenotypic difference between normal astroglia and
evidence for increased microglial activation in the prefrontal
astrocytes affected by the AD pathology. Another kind of
cortex, the motor cortex, the thalamus and the pons of ALS
phenotypic difference was observed in astrocytes from an AD
patients. Both pathologies—the loss of motor neurones and
model expressing double mutated K670N-M671L APP; these
neuroinflammation, can be found in transgenic mice over-
astrocytes began to express β-secretase, thus becoming
expressing mutant variants of the human gene encoding for
possible producers of Aβ (
copper/zinc superoxide dismutase (SOD1), which have been
). While it remains
linked to inherited ALS
unclear to which degree astrocyte activation contributes to Aβ
generation or its clearance, it seems apparent that astrocytes
appearance of activated microglia and astrocytes already at an
contribute to the inflammatory component of AD. For
early, presymptomatic stage of the disease in SOD1 transgenic
example, astrocytes have been shown to express iNOS and
mice suggests that an inflammation may contribute to motor
the L-arginine-supplying enzyme argininosuccinate synthe-
neurones degeneration and the suppression of the inflamma-
tase and consequently contribute to NO- and peroxynitrite
tory component could be neuroprotective. Indeed, increasing
mediated neurotoxicity (
experimental evidence suggests an active and contributory
Although astrocytes serve as a constant and
role of microglia in ALS. Thus, the overexpression of mutant
important source of neurotrophic factors under physiological
human SOD1 in motor neurones alone did not result in
conditions, in vitro and in vivo experiments suggest that
significant neuronal degeneration in transgenic mice (
chronically activated inflammatory astrocytes may not gen-
. A similar approach
erate significant amounts of these molecules (
expressing mutant SOD1 under control of the astrocytic
promoter GFAP, thus causing the astroglial expression of
The AD may also impair other astroglial homeostatic
protein, also failed to induce motor neurones death (
functions. For example, Aβ affects astroglial ability to accu-
In contrast, the selective deletion of mutant SOD1 in
mulate glutamate: treatment of rat cultured astrocytes with
microglia increased the survival rate in the SOD1 transgenic
Aβ1–40 reduced both expression and capacity of GLAST- and
mouse model, indicating that the presence of mutant SOD1 in
GLT-1 mediated glutamate uptake (). Reactive
microglial cells is fundamental for their detrimental effect on
and pathologically changed astrocytes are also responsible for
motor neurone integrity (. The latter
failures in the functional activity of neuronal–glial–vascular
hypothesis was further corroborated by bone marrow trans-
units. Indeed, the vascular dysfunctions, perivascular amiloi-
plantation experiments in mice deficient for myeloid cells but
dosis and compromised blood–brain barrier are inseparable
harbouring the human SOD1 mutant. In these mice, the
parts of AD pathology How astroglial
transplantation of wild type bone marrow cells but not SOD1
cells are participating in these changes remains, however, an
mutant bone marrow cells significantly delayed the progres-
open question.
sion of the disease (
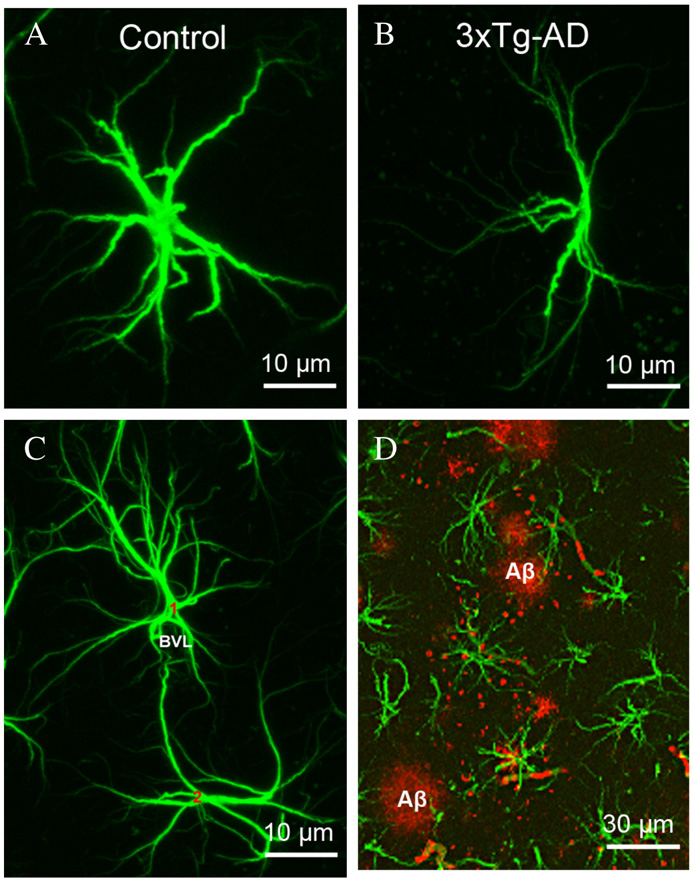
Fig. 3 – Astroglial atrophy and astrogliosis accompany the development of AD pathology in the brains of triple transgenicmouse. (A, B) Representative confocal micrographs illustrating normal control astrocytes (A) compared to the astrocytic atrophyobserved in the dentate gyrus of 3xTg-AD mice (B). The atrophy is manifested by a reduction of the size of somatas as well as inthe reduction of number of primary processes and their branching. (C) Confocal micrograph showing perivascular astrocytes innormal animals. (D) Confocal dual labelling images (GFAP in green and Aβ in red) in 3xTg-AD mice showing the close appositionof astrocytes with the Aβ accumulations. Astrocytes surround Aβ plaques and undergo astrogliosis.
Although the exact molecular mechanism by which micro-
neurones by pioglitazone. This finding was further substanti-
glial cells become activated in ALS remains to be determined,
ated by the superior motor performance in the RotaRod35 and
various approaches targeting the inflammatory component of
grip latency tests of mice treated with pioglitazone. The
the disease demonstrated beneficial effects. Thus, the treat-
numbers of activated microglia were markedly reduced at
ment of mutant SOD1 transgenic mice with minocycline
sites of neurodegeneration in pioglitazone-treated SOD1-G93A
improved motor performance and survival (
mice compared with non-treated mice, as were levels of COX2
). Likewise, the inhibition of COX2 by celecoxib
and iNOS proteins. also provided evidence in a
or nimesulide delayed the onset of the disease
mouse model of ALS that nitric-oxide-dependent peroxynitrite
Using a further anti-inflamma-
generation was reduced by pioglitazone.
tory treatment strategy, two studies with SOD1-G93A trans-genic mice, an established model of ALS, independently found
Parkinson's disease
that oral treatment with the PPARγ agonist pioglitazoneextended the survival of these mice
PD is characterised by a progressive degeneration of dopami-
). Pioglitazone treatment delayed the onset of
nergic midbrain neurones in the substantia nigra (SN) and
disease and prevented a decrease in body weight in these
becomes clinically apparent when more than 50% of SN
mice in comparison with untreated SOD1-G93A mice. The
neurones have been lost. Despite the decades of intensive
quantification of spinal cord motor neurones revealed neuro-
research, the cause of the neurodegeneration in PD is still
protection in mice treated with pioglitazone, whereas non-
poorly understood. Microglial activation has been found in the
treated SOD1-G93A mice had lost 30–40% of these neurones at
substantia nigra (SN) at sites of dopaminergic cell loss in post-
comparable time points. This neuroprotective effect was
mortem human brains derived from PD patients. Likewise,
paralleled by the preservation of the median fibre diameter
activated microglial cells are found in all animal models of PD,
of the quadriceps muscle in treated mice, indicating not only a
together suggesting that neuroinflammatory mechanisms are
functional but also a morphological protection of motor
involved in the disease process. Structurally modified α-
synucleins (α-SYN), particularly nitrated species, which are
are associated with neuritic plaques ()
released as a consequence of dopaminergic neurodegenera-
and they secrete a wide variety of pro-inflammatory molecules
tion, have been found to act as potent microglial immuno-
Furthermore the microglia is
implicated in active phagocytosis of Aβ, thus counterbalancing
Additional experimental evidence leads to the
the Aβ load ). The
hypothesis that neuroinflammation plays an active and
activation of microglia occurs in response to the formation of
promoting role in the disease process based on the finding
neuritic plaques. Several amyloid peptides and APP itself can act
that suppression of inflammatory signalling cascades sub-
as potent glial activators
stantially improved the phenotypic outcome as well as
), whereas the disruption of the
protected from dopaminergic cell loss and subsequent neuro-
APP gene and its proteolytic products delay and decrease
chemical changes in SN projection areas. Thus, iNOS inhibi-
microglial activation ). Microglial cells
tion by either genetic deficiency or pharmacological treatment
have been suggested to be preferentially associated with certain
has been found to exert neuroprotection
amyloid plaque types indicating that plaque development and
). Similarly, more general anti-inflam-
the degree of microglial reaction are interrelated
matory approaches including the activation of the peroxisome
). However, it remains unclear whether Aβ plaque deposi-
proliferator activated receptor gamma pioglitazone
tion is an absolute requirement for microglial activation, or
) or treatment with the semi-
whether this can already be evoked by soluble and toxic Aβ
synthetic tetracycline minocyline significantly protected from
species. This hypothesis is supported by a recent study where
dopaminergic neurodegeneration. All of these experimental
the focal activation of microglial cells becomes apparent at
approaches aim to block or at least interact with mechanisms
3 months of age in APP V717I transgenic mice, which usually
that finally execute neuronal cell death such as oxidative
start to deposit Aβ in plaque like structures much later—at
stress and cytokine-receptor-mediated apoptosis.
around 10–12 months (). In contrast, studies
Microglia may, however, have also protective functions in
using in vivo multiphoton microscopy using 5–6 month old B6C3-
this disease, for example by secretion of anti-inflammatory and
YFP transgenic mice (bearing APPswe and PS1d9x-YFP genes)
neuroprotective cytokines such as TGF-β (). An
suggested that microglial are recruited to Aβ plaques only after
important question is, as to what extent the local inflammatory
they have been formed ).
process within the substantia nigra can be influenced by
The mechanisms of microglial activation by Aβ depositions
peripheral inflammatory events. In a recent study Godoy et al.
are not yet clear, although several receptor systems are directly
tested whether a sub-toxic dose of bacterial lipopolysaccharide
implicated in this process. In particular, the activation of
(LPS) is actively modulating microglia from an anti- to a pro-
microglia requires P2X7 purinoceptors and Ca2+ signalling. The
inflammatory state and thereby exacerbate disease progression.
exposure of cultured microglial cells to Aβ25–35 triggers Ca2+
In this experiment, LPS injection in the degenerating SN
influx, the IL-1β release and P2X7-dependent membrane
exacerbated neurodegeneration, worsened the behavioural
permeabilisation, all being absent in cells prepared from P2X7
phenotype and caused an increase in microglial IL-1 secretion.
KO mice (). Furthermore, the intra-hippocam-
Of note, IL-1 inhibition reversed these effects. Importantly,
pal injection of Aβ1–42 failed to induce microglial activation (as
chronic systemic IL-1 also exacerbated neurodegeneration and
judged by IL-1β accumulation) in animals deficient in P2X7
microglial activation in the SN (Interestingly,
in a very recent paper, Smeyne et al. showed that abdominal
The activation of microglial cells by aggregated Aβ involves
infection with the H5N1 influenza virus resulted in a very rapid
Toll-like receptors (of TLR4 type; the TLR4
viral migration into the CNS and viral presence predominantly
receptors are up-regulated in both AD brain preparations and
within brainstem and midbrain nuclei (). Viral
in APP transgenic mice. A spontaneous loss-of-function
neurotrophism caused a robust inflammatory reaction within
mutation in the TLR4 gene significantly reduced Aβ-induced
these nuclei including the locus coeruleus and SN. Importantly,
microglial activation Exposure of micro-
the H5N1 virus induced neuroinflammation, which persisted
glial cultures to Aβ also stimulated TLR2 receptors, while
beyond the presence of the virus itself and caused α-SYN
inhibiting TLR9 receptors ). The stimulation of
aggregation and progressive neurodegeneration. Together,
the TLR-associated signalling system may have dual effect in
these data suggest that the brainstem nuclei such as the SN
AD progression. On one hand, the activation of TLRs increases
are vulnerable to peripheral infection and immunological
microglial phagocytosis of Aβ (this involves p38 MAPK
processes. In keeping with this, anti-inflammatory therapeutic
signalling and expression of G-protein-coupled formyl peptide
strategies that target proinflammatory microglial activation
receptor-like 2, mFPR2; the latter likely being the sensor for Aβ
may represent a future therapeutic avenue.
Atthe same time, however, the over-stimulation of TLRs may
Alzheimer's disease
trigger excessive release of cytokines, proteases and othercytotoxins thus promoting neural cell death ().
Next to the classical neuropathological features of AD, namely
The Aβ stimulates a nuclear factor kappa B (NFκB)-depen-
Aβ deposition and neurofibrillary tangle formation, neuroin-
dent pathway that is required for cytokine gene transcription
flammatory changes have been identified as the third important
within activated microglia and reactive
component of the disease. The inflammatory reactions of
astrocytes. Not only Aβ, but also the carboxy-terminal 100
microglia and astroglia are intimately associated with the
amino acids of APP (CT100) (which are present in senile plaques)
pathogenesis and progress of AD. The activated microglial cells
can induce astrogliosis and neuronal death. Exposure to CT100
results in activation of the mitogen-activated protein kinase
Thus, demonstrated that
(MAPK) pathways as well as NFκB ). Addition-
complement activation can protect against Aβ-induced toxic-
ally, other proteins involved in APP processing have been
ity and may reduce the accumulation or promote the
implicated in the inflammatory response. Loss of presenilin
clearance of senile plaques. The AD mice expressing a soluble
function in presenilin conditional knockout mice leads to
form of the complement inhibitor Crry, which blocks C3
differential up-regulation of inflammatory markers in the
activation, under the control of the glial fibrillary acidic
cerebral cortex, such as strong microglial activation, comple-
protein promoter displayed higher Aβ deposition and more
ment component C1q, and cathepsin S (
prominent neurodegeneration than age-matched control
Once stimulated, microglia participate in the generation and
mice. However, more recently it was reported that transgenic
release of a wide range of inflammatory mediators including
mouse models of AD lacking C1q showed reduced pathology,
complement factors, chemokines and cytokines. The comple-
consisting of decreased numbers of activated microglia and
ment system represents a complex and tightly regulated attack
improved neuronal integrity, without changes in plaque area.
cascade designed to destroy invaders and assist in the
These data suggest that at stages when fibrillar plaque
phagocytosis of waste, one of the key microglial tasks under
pathology is present, C1q exerts a detrimental effect on
physiological and pathophysiological conditions. The compo-
neuronal integrity, most likely through the activation of the
nents of this system carry out four major functions: recogni-
classical complement cascade.
tion, opsonisation, inflammatory stimulation and direct killing
In AD, unlike in the aforementioned neurological disorders
through the membrane attack complex (MAC)
characterised by leukocyte infiltration, abnormal or excessive
In addition to triggering the generation of a
migration of inflammatory cells into the CNS has not been
membranolytic complex, complement proteins interact with
definitively shown to occur. Nonetheless, there is growing
cell surface receptors to promote a local inflammatory response
evidence that chemokines and chemokine receptors are up-
that contributes to the protection and healing of the host.
regulated in resident CNS cells in an AD brain
Microglial complement activation causes inflammation and
), and chemokines may contribute to plaque-
cell damage, yet it is essential for eliminating cell debris and
associated inflammation and neurodegeneration. Up-regula-
potentially toxic protein aggregates. The complement system
tion of CXCR2 expression has been observed on some
consists of some 30 fluid-phase and cell-membrane associated
dystrophic neurites in senile plaques (
proteins that can be activated by three different routes. The
). In addition, the expression of CCR3 and
classical pathway (involving C1q, C1r, C1s, C4, C2, and C3
CCR5 is increased on some reactive microglia in AD, and MIP-
components) is activated primarily by the interaction of C1q
1α is found in a subpopulation of reactive astrocytes (
with immune complexes (antibody–antigen), but activation
MCP-1 has also been localised to mature senile plaques
can also be achieved after the interaction of C1q with non-
and reactive microglia, but is not found in immature senile
immune molecules such as DNA, RNA, C-reactive protein,
plaques. Furthermore, in vitro studies have demonstrated the
serum amyloid P, bacterial lipopolysaccharides, some fungal
ability of Aβ to stimulate the production of IL-8, MCP-1, MIP-1α
and virus membranes, and most importantly, by fibrillar Aβ.
and MIP-1β from human monocytes (). For
The initiation of the alternative pathway (involving C3, factor
example, microglia cultured from rapid autopsies of AD and
B, factor D, and properdin) does not require the presence of
non-demented patients exhibit significant, dose-dependent
immune complexes and leads to the deposition of C3 frag-
increases in IL-8, MCP-1 and MIP-1α after an exposure to Aβ
ments on target cells. Mannose-binding lectin (MBL), a lectin
Although more studies are certainly needed,
homologous to C1q, can recognise carbohydrates such as
it is likely that plaque-associated chemokine production plays
mannose and N-acetylglucosamine on pathogens and initiate
a role in the recruitment and accumulation of microglia to the
the complement pathway independently of both the classical
neuritic plaques. Future studies using targeted disruption of
and the alternative activation pathways. Like the C1 complex
chemokines and chemokine receptors in mouse models of AD
in the classical pathway, MBL is associated with two serine
should help to clarify the role of chemokines in plaque-
proteases that cleave C4 and C2 components, leading to the
associated inflammation and neurodegeneration.
formation of the classical C3 convertase (
Microglia derived cytokines associated with AD include
Microglial cells can produce complement proteins to
several interleukins (ILs), TNF-α and TGFβ amongst others. In
recognise and kill pathogens locally. Studies using RT-PCR
general, cytokine production is increased in inflammatory
have shown locally up-regulated complement mRNA in AD
states and they function by regulating the intensity and
brain, especially in the areas of primary pathology: the
duration of the immune response
entorhinal cortex, the hippocampus, and the midtemporal
). Thus, IL-1 induces IL-6 produc-
gyrus Numerous groups have reported
tion, stimulates iNOS activity ), and
the association of complement proteins of the classical
induces the production of M-CSF
pathway, particularly the MAC, with amyloid plaques and
In addition, IL-1 enhances neuronal
neurofibrillary tangles in AD brains (
acetylcholinesterase activity, microglial activation and addi-
Information about the functional role comes from studies of
tional glial IL-1 production, with consequent activation, and
mutant mice lacking complement proteins, which suggest
expression of the cytokine S100β by astrocytes, thereby
that impaired phagocytosis can result in immunomediated
establishing a self-propagating cycle (
tissue damage and inflammation
). The IL-6 promotes astrogliosis
). However, the complement system may be Janus-faced
activates microglia and stimulates
and also provide beneficial action to the brain during AD.
the production of acute phase proteins (). The
knockout mice deficient in IL-6 exhibit a facilitation of radial
enzymes regulate the generation of a whole spectrum of
maze learning over 30 days and show a faster acquisition,
prostanoids, some of which may be neuroprotective and others
suggesting a possible negative regulation of memory formation
neurodestructive. Thus, the composition and proportion of all
and consolidation processes by IL-6 (). TNF-α
prostanoids together may actually determine whether the
has both pro-apoptotic and anti-apoptotic effects. This proin-
activity of COX enzymes is beneficial or detrimental.
flammatory cytokine accounts for most of the neurotoxic
In vitro, LPS activated microglial cells and IL-1β-stimulated
activity secreted by monocytes and microglia
astroglial cells are capable of synthesising COX-2
). On the other hand, TNF-α has been reported to have
). In contrast to
neuroprotective properties ) in the AD brain.
peripheral monocytes, cultured rat microglia cells do not
In addition to the general role of cytokines, AD-specific
synthesise COX-2 in response to IL-1 or IL-6 ),
interactions of certain cytokines with the APP processing
suggesting that COX-2 regulation differs between CNS and
pathway and Aβ may be pathophysiologically relevant. For
peripheral cells. In rat microglial cell cultures, the major
example, IL-1 can regulate APP processing and Aβ production in
enzymatic product of COX-2 appears to be prostaglandin E2
vitro ). In turn, fibrillar Aβ has been reported to
(PGE2). Because PGE2 itself is able to induce COX-2 in microglia
increase neurotoxic secretory products, proinflammatory cyto-
), some sort of autocrine or paracrine
kines and reactive oxygen species (
amplification of the COX-2 induction in microglial cells or a
spreading of the COX-2 expression between neurones and
Cultured rat cortical glia exhibit elevated IL-6 mRNA after
microglial cells is possible. The PGE2 acts on four different
exposure to the carboxy-terminal 105 amino acids of APP
receptors designated as EP1 to EP4 EP1
(). Incubation of cultured microglia with Aβ
and EP2 receptors have been detected in cultured microglia,
increased IL-1, IL-6, TNF-α MIP-1α and MCP-1 in a dose-
while EP3 receptors are also present in activated microglia in
dependent manner (
vivo ). Microglial EP2 receptors inhibit
phagocytosis and enhance neurotoxic activities of microglia
Altogether, Aβ stimulated produc-
). PGE2 may also act on the neuronal EP2
tion of interleukins and other cytokines and chemokines and
receptor, which is involved in apoptosis, although investiga-
their feedback activation of APP production or BACE1
tions of the role of EP2 activation on neuronal cell death have
) may establish a self-perpetuating, vicious
yielded conflicting results and somewhat suggested a neuro-
cycle. A second general category of cytokine action is mani-
protective role of neuronal EP2 stimulation under several
fested by inhibitory, anti-inflammatory cytokines such as IL-1
pathophysiological circumstances (
receptor antagonist (IL-1Ra), IL-4, IL-10 and TGF-β. Some of
these are reportedly elevated in AD, consistent with induction of
This is further exemplified in a
homeostatic mechanisms in neuroinflammation
recent report where the knockout of EP2 in a double transgenic
(APP/PS1) mouse led to decreased evidence of oxidative stress
use of anti-inflammatory cytokines such as IL-4 and TGF-β
and decreased Aβ production, associated with lower levels of
could be beneficial, because they are able to inhibit CD40 and
BACE (). In conclusion, the neuronal and glial
class II MHC by restricting their expression and activity
secretion of PGE2 may impair the phagocytotic clearance of Aβ
). However, an overexpression of TGF-β
by binding to the microglia EP2 receptor and enhancing
in transgenic mice leads to changes in the microvasculature,
microglial toxicity. However the role of PGE2 in neurodegenera-
including age related amyloid deposition
tion may be far more complex due to the presence of other EP
reflecting the multi-functional nature of many cytokines.
receptor subtypes on microglial cells and the effects of PGE2 on
In addition to the above described evidence from the analysis of
other cell types. Neuronal death elicited by excitotoxins is
human brain tissue, cell culture and transgenic animal studies,
elevated in transgenic animals with high expression of COX-2,
an association of AD with several polymorphisms of proin-
suggesting that the COX-2 expression may further interact with
flammatory genes has been described, including IL-1 (
other pathogenetic mechanisms
), IL-6 ), TNF-α
It should be noted that some aspects of microglial function
and α1-antichymotrypsin, an acute
may be beneficial, since activated microglia are able to reduce
phase protein ). However, none of the
Aβ accumulation by increasing its phagocytosis, clearance
various members of the interleukin cytokine family that are
and degradation ). Thus,
associated with AD actually map to chromosomal regions with
secreted Aβ1–40 and Aβ1–42 peptides are constitutively degrad-
evidence of genetic linkage ). Thus,
ed by the insulin degrading enzyme (IDE), a metalloprotease
although inflammation and the up-regulation of inflammatory
released by microglia and other neural cells. Finally, microglia
mediators like the interleukins are regularly observed in AD
can also secrete several trophic factors, such as the glia-
brain, it appears less likely that variation at the genomic level of
derived neurotrophic factor (GDNF), which exert a well
these proteins makes a large contribution to AD risk in general.
documented neuroprotective function (
Next to the complement factors, chemokines and cytokines,
activated microglia can also serve as a chief source ofprostanoids. Two isoforms of cyclooxygenases, the mainly
Concluding remarks
constitutively expressed COX-1 and the inducible COX-2,catalyze key steps of prostanoid synthesis in mammalian
The neurodegenerative diseases result from the failure in
cells. Downstream of both COX-1 and COX-2 several other
brain connectivity, which is formed by neuronal–neuronal,
neuronal–glial and glial–glial contacts. Contrary to past
pro-inflammatory enzyme cyclooxygenase-2 in amyotrophic
beliefs, which regarded neuropathology to be solely associated
lateral sclerosis. Ann. Neurol. 49, 176–185.
Aloisi, F., Care, A., Borsellino, G., Gallo, P., Rosa, S., Bassani, A.,
with neuronal malfunction and neuronal cell death, the new
Cabibbo, A., Testa, U., Levi, G., Peschle, C., 1992. Production of
doctrine, which regards glia as a central element in neuro-
hemolymphopoietic cytokines (IL-6, IL-8, colony-stimulating
logical diseases is emerging. The neurodegeneration is driven
factors) by normal human astrocytes in response to IL-1-β and
by complex astroglial reactions which include astroglial
tumor-necrosis-factor-α. J. Immunol. 149, 2358–2366.
atrophy with a subsequent impairment of synaptic transmis-
Alzheimer, A., 1910. Beiträge zur Kenntnis der pathologischen
sion and astrogliosis, which control both neuroprotective and
Neuroglia und ihrer Beziehungen zu den Abbauvorgängen im
neurotoxic reactions. The activation of microglia, associated
Nervengewebe. In: Nissl, F., Alzheimer, A. (Eds.), Histologischeund histopathologische Arbeiten über die Grosshirnrinde mit
with the secretion of pro-inflammatory factors further con-
besonderer Berücksichtigung der pathologischen Anatomie
tributes to regulation of neuroprotection/neurotoxic balance.
der Geisteskrankheiten. Gustav Fischer, Jena, pp. 401–562.
This recently acquired knowledge allows us to regard the
Andriezen, W.L., 1893. The neuroglia elements of the brain. Brit.
neurodegenerative diseases as primarily gliodegenerative
Med. J. 2, 227–230.
processes, in which glial cells determine the progression and
Angulo, M.C., Le Meur, K., Kozlov, A.S., Charpak, S., Audinat, E.,
outcome of neuropathological process.
2008. GABA, a forgotten gliotransmitter. Prog. Neurobiol. 86,297–303.
Antel, J., Arnold, D., 2005. Multiple sclerosis. In: Kettenmann, H.,
Ransom, B. (Eds.), Neuroglia. Oxford University Press, Oxford,
pp. 489–500.
Araque, A., Parpura, V., Sanzgiri, R.P., Haydon, P.G., 1999. Tripartite
Authors research was supported by Deutsche Forschungsge-
synapses: glia, the unacknowledged partner. Trends Neurosci.
meinschaft, Deutsche Krebshilfe (10-6789) and the Wilhelm
22, 208–215.
Bach, J.H., Chae, H.S., Rah, J.C., Lee, M.W., Park, C.H., Choi, S.H.,
Sander foundation (2006.001.1) grants to MTH; Alzheimer's
Choi, J.K., Lee, S.H., Kim, Y.S., Kim, K.Y., Lee, W.B., Suh, Y.H.,
Research Trust (UK) Programme Grant (ART/PG2004A/1) to AV
Kim, S.S., 2001. C-terminal fragment of amyloid precursor
and JJR; National Institute of Health (NIH) grant to AV, Grant
protein induces astrocytosis. J. Neurochem. 78, 109–120.
Agency of the Czech Republic (GACR 309/09/1696) to JJR and
Barbeito, L.H., Pehar, M., Cassina, P., Vargas, M.R., Peluffo, H., Viera,
(GACR 305/08/1384) to AV.
L., Estevez, A.G., Beckman, J.S., 2004. A role for astrocytes inmotor neuron loss in amyotrophic lateral sclerosis. Brain Res.
Brain Res. Rev. 47, 263–274.
Barger, S.W., Harmon, A.D., 1997. Microglial activation by
Alzheimer amyloid precursor protein and modulation byapolipoprotein E. Nature 388, 878–881.
Abbracchio, M.P., Burnstock, G., Verkhratsky, A., Zimmermann, H.,
Bauer, M.K., Lieb, K., Schulze-Osthoff, K., Berger, M., Gebicke-Haerter,
2009. Purinergic signalling in the nervous system: an overview.
P.J., Bauer, J., Fiebich, B.L., 1997. Expression and regulation
Trends Neurosci. 32, 19–29.
of cyclooxygenase-2 in rat microglia. Eur. J. Biochem. 243,
Abramov, A.Y., Canevari, L., Duchen, M.R., 2003. Changes in
intracellular calcium and glutathione in astrocytes as the
Beers, D.R., Henkel, J.S., Xiao, Q., Zhao, W.H., Wang, J.H., Yen, A.A.,
primary mechanism of amyloid neurotoxicity. J. Neurosci. 23,
Siklos, L., McKercher, S.R., Appel, S.H., 2006. Wild-type
microglia extend survival in PU.1 knockout mice with familial
Abramov, A.Y., Canevari, L., Duchen, M.R., 2004. β-Amyloid
amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. U.S.A. 103,
peptides induce mitochondrial dysfunction and oxidative
stress in astrocytes and death of neurons through activation of
Beglopoulos, V., Sun, X., Saura, C.A., Lemere, C.A., Kim, R.D., Shen,
NADPH oxidase. J. Neurosci. 24, 565–575.
J., 2004. Reduced beta-amyloid production and increased
Agulhon, C., Petravicz, J., McMullen, A.B., Sweger, E.J., Minton, S.K.,
inflammatory responses in presenilin conditional knock-out
Taves, S.R., Casper, K.B., Fiacco, T.A., McCarthy, K.D., 2008.
mice. J. Biol. Chem. 279, 46907–46914.
What is the role of astrocyte calcium in neurophysiology?
Bell, R.D., Zlokovic, B.V., 2009. Neurovascular mechanisms and
Neuron 59, 932–946.
blood–brain barrier disorder in Alzheimer's disease. Acta
Akiguchi, I., Tomimoto, H., Suenaga, T., Wakita, H., Budka, H.,
Neuropathol. 118, 103–113.
1997. Alterations in glia and axons in the brains of
Benveniste, E.N., Nguyen, V.T., O'Keefe, G.M., 2001. Immunological
Binswanger's disease patients. Stroke 28, 1423–1429.
aspects of microglia: relevance to Alzheimer's disease.
Akiyama, H., Barger, S., Barnum, S., Bradt, B., Bauer, J., Cole, G.M.,
Neurochem. Int. 39, 381–391.
Cooper, N.R., Eikelenboom, P., Emmerling, M., Fiebich, B.L.,
Bergles, D.E., Roberts, J.D., Somogyi, P., Jahr, C.E., 2000.
Finch, C.E., Frautschy, S., Griffin, W.S., Hampel, H., Hull, M.,
Glutamatergic synapses on oligodendrocyte precursor cells in
Landreth, G., Lue, L., Mrak, R., Mackenzie, I.R., McGeer, P.L.,
the hippocampus. Nature 405, 187–191.
O'Banion, M.K., Pachter, J., Pasinetti, G., Plata-Salaman, C.,
Berry, M., Hubbard, P., Butt, A.M., 2002. Cytology and lineage of
Rogers, J., Rydel, R., Shen, Y., Streit, W., Strohmeyer, R.,
NG2-positive glia. J. Neurocytol. 31, 457–467.
Tooyoma, I., Van Muiswinkel, F.L., Veerhuis, R., Walker, D.,
Bezzi, P., Carmignoto, G., Pasti, L., Vesce, S., Rossi, D., Rizzini, B.L.,
Webster, S., Wegrzyniak, B., Wenk, G., Wyss-Coray, T., 2000.
Pozzan, T., Volterra, A., 1998. Prostaglandins stimulate
Inflammation and Alzheimer's disease. Neurobiol. Aging 21,
calcium-dependent glutamate release in astrocytes. Nature
391, 281–285.
Almer, G., Vukosavic, S., Romero, N., Przedborski, S., 1999.
Bezzi, P., Domercq, M., Brambilla, L., Galli, R., Schols, D., De Clercq,
Inducible nitric oxide synthase up-regulation in a transgenic
E., Vescovi, A., Bagetta, G., Kollias, G., Meldolesi, J., Volterra, A.,
mouse model of familial amyotrophic lateral sclerosis. J.
2001. CXCR4-activated astrocyte glutamate release via TNFα:
Neurochem. 72, 2415–2425.
amplification by microglia triggers neurotoxicity. Nat.
Almer, G., Guegan, C., Teismann, P., Naini, A., Rosoklija, G., Hays,
Neurosci. 4, 702–710.
A.P., Chen, C., Przedborski, S., 2001. Increased expression of the
Bezzi, P., Gundersen, V., Galbete, J.L., Seifert, G., Steinhauser, C.,
Pilati, E., Volterra, A., 2004. Astrocytes contain a vesicular
receptor 2 on microglia promotes cell uptake of Alzheimer
compartment that is competent for regulated exocytosis of
disease-associated amyloid beta peptide. J. Biol. Chem. 281,
glutamate. Nat. Neurosci. 7, 613–620.
Biber, K., Neumann, H., Inoue, K., Boddeke, H.W., 2007. Neuronal
Chittajallu, R., Aguirre, A., Gallo, V., 2004. NG2-positive cells in the
‘on' and ‘off' signals control microglia. Trends Neurosci. 30,
mouse white and grey matter display distinct physiological
properties. J. Physiol. 561, 109–122.
Bilak, M., Wu, L., Wang, Q., Haughey, N., Conant, K., St Hillaire, C.,
Chong, Y., 1997. Effect of a carboxy-terminal fragment of the
Andreasson, K., 2004. PGE2 receptors rescue motor neurons in a
Alzheimer's amyloid precursor protein on expression of
model of amyotrophic lateral sclerosis. Ann. Neurol. 56, 240–248.
proinflammatory cytokines in rat glial cells. Life Sci. 61,
Blasko, I., Marx, F., Steiner, E., Hartmann, T., Grubeck-Loebenstein,
B., 1999. TNFα plus IFNγ induce the production of Alzheimer
Christopherson, K.S., Ullian, E.M., Stokes, C.C., Mullowney, C.E.,
β-amyloid peptides and decrease the secretion of APPs. FASEB
Hell, J.W., Agah, A., Lawler, J., Mosher, D.F., Bornstein, P.,
J. 13, 63–68.
Barres, B.A., 2005. Thrombospondins are astrocyte-secreted
Blumenthal, I., 2004. Periventricular leucomalacia: a review. Eur. J.
proteins that promote CNS synaptogenesis. Cell 120, 421–433.
Pediatr. 163, 435–442.
Cohen-Gadol, A.A., Pan, J.W., Kim, J.H., Spencer, D.D., Hetherington,
Boillee, S., Yamanaka, K., Lobsiger, C.S., Copeland, N.G., Jenkins,
H.H., 2004. Mesial temporal lobe epilepsy: a proton magnetic
N.A., Kassiotis, G., Kollias, G., Cleveland, D.W., 2006. Onset and
resonance spectroscopy study and a histopathological analysis.
progression in inherited ALS determined by motor neurons
J. Neurosurg. 101, 613–620.
and microglia. Science 312, 1389–1392.
Coles, J.A., Orkand, R.K., 1983. Modification of potassium move-
Bolmont, T., Haiss, F., Eicke, D., Radde, R., Mathis, C.A., Klunk, W.E.,
ment through the retina of the drone (Apis mellifera male) by
Kohsaka, S., Jucker, M., Calhoun, M.E., 2008. Dynamics of the
glial uptake. J. Physiol. 340, 157–174.
microglial/amyloid interaction indicate a role in plaque
Combs, C.K., Bates, P., Karlo, J.C., Landreth, G.E., 2001a. Regulation
maintenance. J. Neurosci. 28, 4283–4292.
of β-amyloid stimulated proinflammatory responses by
Botto, M., 1998. C1q knock-out mice for the study of complement
peroxisome proliferator-activated receptor alpha. Neurochem.
deficiency in autoimmune disease. Exp. Clin. Immunogenet.
Int. 39, 449–457.
15, 231–234.
Combs, C.K., Karlo, J.C., Kao, S.C., Landreth, G.E., 2001b. β-Amyloid
Braida, D., Sacerdote, P., Panerai, A.E., Bianchi, M., Aloisi, A.M.,
stimulation of microglia and monocytes results in TNFα-
Iosue, S., Sala, M., 2004. Cognitive function in young and adult
dependent expression of inducible nitric oxide synthase and
IL (interleukin)-6 deficient mice. Behav. Brain Res. 153, 423–429.
neuronal apoptosis. J. Neurosci. 21, 1179–1188.
Breidert, T., Callebert, J., Heneka, M.T., Landreth, G., Launay, J.M.,
Cornell Bell, A.H., Finkbeiner, S.M., Cooper, M.S., Smith, S.J., 1990.
Hirsch, E.C., 2002. Protective action of the peroxisome
Glutamate induces calcium waves in cultured astrocytes:
proliferator-activated receptor-gamma agonist pioglitazone
long-range glial signaling. Science 247, 470–473.
in a mouse model of Parkinson's disease. J. Neurochem. 82,
Dabir, D.V., Trojanowski, J.Q., Richter-Landsberg, C., Lee, V.M.,
Forman, M.S., 2004. Expression of the small heat-shock protein
Broe, M., Kril, J., Halliday, G.M., 2004. Astrocytic degeneration
αB-crystallin in tauopathies with glial pathology. Am. J. Pathol.
relates to the severity of disease in frontotemporal dementia.
164, 155–166.
Brain 127, 2214–2220.
Danbolt, N.C., 2001. Glutamate uptake. Progr. Neurobiol. 65, 1–105.
Bruzzone, R., Giaume, C., 1999. Connexins and information
D'Andrea, M.R., Cole, G.M., Ard, M.D., 2004. The microglial
transfer through glia. Adv. Exp. Med. Biol. 468, 321–337.
phagocytic role with specific plaque types in the Alzheimer
Bushong, E.A., Martone, M.E., Jones, Y.Z., Ellisman, M.H., 2002.
disease brain. Neurobiol. Aging 25, 675–683.
Protoplasmic astrocytes in CA1 stratum radiatum occupy
Dani, J.W., Chernjavsky, A., Smith, S.J., 1992. Neuronal activity
separate anatomical domains. J. Neurosci. 22, 183–192.
triggers calcium waves in hippocampal astrocyte networks.
Bushong, E.A., Martone, M.E., Ellisman, M.H., 2004. Maturation of
Neuron 8, 429–440.
astrocyte morphology and the establishment of astrocyte
Davalos, D., Grutzendler, J., Yang, G., Kim, J.V., Zuo, Y., Jung, S.,
domains during postnatal hippocampal development. Int. J.
Littman, D.R., Dustin, M.L., Gan, W.B., 2005. ATP mediates rapid
Dev. Neurosci. 22, 73–86.
microglial response to local brain injury in vivo. Nat. Neurosci.
Butovsky, O., Talpalar, A.E., Ben Yaakov, K., Schwartz, M., 2005.
8, 752–758.
Activation of microglia by aggregated β-amyloid or
DeGiorgio, L.A., Shimizu, Y., Chun, H.S., Cho, B.P., Sugama, S., Joh,
lipopolysaccharide impairs MHC-II expression and renders
T.H., Volpe, B.T., 2002. APP knockout attenuates microglial
them cytotoxic whereas IFN-γ and IL-4 render them protective.
activation and enhances neuron survival in substantia nigra
Mol. Cell Neurosci. 29, 381–393.
compacta after axotomy. Glia 38, 174–178.
Butt, A.M., 2005. Structure and function of oligodendrocytes. In:
Dehmer, T., Lindenau, J., Haid, S., Dichgans, J., Schulz, J.B., 2000.
Kettenmann, H., Ransom, B. (Eds.), Neuroglia. Oxford
Deficiency of inducible nitric oxide synthase protects against
University Press, Oxford, pp. 36–47.
MPTP toxicity in vivo. J. Neurochem. 74, 2213–2216.
Butt, A.M., Hamilton, N., Hubbard, P., Pugh, M., Ibrahim, M., 2005.
Dehmer, T., Heneka, M.T., Sastre, M., Dichgans, J., Schulz, J.B., 2004.
Synantocytes: the fifth element. J. Anat. 207, 695–706.
Protection by pioglitazone in the MPTP model of Parkinson's
Castell, J.V., Gomez-Lechon, M.J., David, M., Andus, T., Geiger, T.,
disease correlates with I kappa B alpha induction and block
Trullenque, R., Fabra, R., Heinrich, P.C., 1989. Interleukin-6 is
of NF kappa B and iNOS activation. J. Neurochem. 88,
the major regulator of acute phase protein synthesis in adult
human hepatocytes. FEBS Lett. 242, 237–239.
Deitmer, J.W., Verkhratsky, A.J., Lohr, C., 1998. Calcium signalling
Charcot, J.M., 1881. Amyotrophic lateral sclerosis: symptomatology.
in glial cells. Cell. Calc. 24, 405–416.
Lectures on diseases of the nervous system. New Sydenham
Dermietzel, R., Gao, Y., Scemes, E., Vieira, D., Urban, M., Kremer,
Society, London, pp. 192–204.
M., Bennett, M.V., Spray, D.C., 2000. Connexin43 null mice
Charcot, J.M., Joffroy, A., 1869. Deux cas d'atrophie musculaire
reveal that astrocytes express multiple connexins. Brain Res.
progressive avec lesions de la substance grise et de faisceaux
Brain Res. Rev. 32, 45–56.
anterolateraux de la moelle epiniere. Arch. Physiol. Norm.
Deshpande, M., Zheng, J., Borgmann, K., Persidsky, R., Wu, L.,
Pathol. 1, 354–367.
Schellpeper, C., Ghorpade, A., 2005. Role of activated astrocytes
Chen, K., Iribarren, P., Hu, J., Chen, J., Gong, W., Cho, E.H., Lockett,
in neuronal damage: potential links to HIV-1-associated
S., Dunlop, N.M., Wang, J.M., 2006. Activation of Toll-like
dementia. Neurotox. Res. 7, 183–192.
DeWitt, D.A., Perry, G., Cohen, M., Doller, C., Silver, J., 1998.
Golgi, C., 1903. Opera Omnia. Milano, Hoepli.
Astrocytes regulate microglial phagocytosis of senile plaque
Gong, Y.H., Parsadanian, A.S., Andreeva, A., Snider, W.D., Elliott,
cores of Alzheimer's disease. Exp. Neurol. 149, 329–340.
J.L., 2000. Restricted expression of G86R Cu/Zn superoxide
Dickson, D.W., Lee, S.C., Mattiace, L.A., Yen, S.H., Brosnan, C., 1993.
dismutase in astrocytes results in astrocytosis but does not
Microglia and cytokines in neurological disease, with special
cause motoneuron degeneration. J. Neurosci. 20, 660–665.
reference to AIDS and Alzheimer's disease. Glia 7, 75–83.
Grammas, P., Ovase, R., 2001. Inflammatory factors are elevated in
Di Giorgio, F.P., Carrasco, M.A., Siao, M.C., Maniatis, T., Eggan, K.,
brain microvessels in Alzheimer's disease. Neurobiol. Aging 22,
2007. Non-cell autonomous effect of glia on motor neurons in
an embryonic stem cell-based ALS model. Nat. Neurosci. 10,
Griffin, W.S.T., 2000. IL-1 and the cytokine cycle in Alzheimer's
disease. J. Neurochem. 74, S52.
Drachman, D.B., Frank, K., Dykes-Hoberg, M., Teismann, P., Almer,
Gurnett, C.A., Landt, M., Wong, M., 2003. Analysis of cerebrospinal
G., Przedborski, S., Rothstein, J.D., 2002. Cyclooxygenase 2
fluid glial fibrillary acidic protein after seizures in children.
inhibition protects motor neurons and prolongs survival
Epilepsia 44, 1455–1458.
in a transgenic mouse model of ALS. Ann. Neurol. 52,
Gurney, M.E., Pu, H., Chiu, A.Y., Dal Canto, M.C., Polchow, C.Y.,
Alexander, D.D., Caliendo, J., Hentati, A., Kwon, Y.W., Deng, H.X.,
Duan, S., Anderson, C.M., Keung, E.C., Chen, Y., Swanson, R.A.,
1994. Motor neuron degeneration in mice that express a human
2003. P2X7 receptor-mediated release of excitatory amino acids
Cu,Zn superoxide dismutase mutation. Science 264,
from astrocytes. J. Neurosci. 23, 1320–1328.
Eid, T., Lee, T.S., Thomas, M.J., Amiry-Moghaddam, M., Bjornsen,
Halassa, M.M., Fellin, T., Haydon, P.G., 2007a. The tripartite
L.P., Spencer, D.D., Agre, P., Ottersen, O.P., de Lanerolle, N.C.,
synapse: roles for gliotransmission in health and disease.
2005. Loss of perivascular aquaporin 4 may underlie deficient
Trends Mol. Med. 13, 54–63.
water and K+ homeostasis in the human epileptogenic
Halassa, M.M., Fellin, T., Takano, H., Dong, J.H., Haydon, P.G.,
hippocampus. Proc. Natl. Acad. Sci. U. S. A. 102, 1193–1198.
2007b. Synaptic islands defined by the territory of a single
Eikelenboom, P., van Gool, W.A., 2004. Neuroinflammatory
astrocyte. J. Neurosci. 27, 6473–6477.
perspectives on the two faces of Alzheimer's disease. J. Neural.
Hanisch, U.K., 2002. Microglia as a source and target of cytokines.
Transm. 111, 281–294.
Glia 40, 140–155.
Eikelenboom, P., Zhan, S.S., van Gool, W.A., Allsop, D., 1994.
Hanisch, U.K., Kettenmann, H., 2007. Microglia: active sensor and
Inflammatory mechanisms in Alzheimer's disease. Trends
versatile effector cells in the normal and pathologic brain. Nat.
Pharmacol. Sci. 15, 447–450.
Neurosci. 10, 1387–1394.
Ercolini, A.M., Miller, S.D., 2006. Mechanisms of immunopathology
Hartlage-Rubsamen, M., Zeitschel, U., Apelt, J., Gartner, U., Franke,
in murine models of central nervous system demyelinating
H., Stahl, T., Gunther, A., Schliebs, R., Penkowa, M., Bigl, V.,
disease. J. Immunol. 176, 3293–3298.
Rossner, S., 2003. Astrocytic expression of the Alzheimer's
Fellin, T., Pascual, O., Gobbo, S., Pozzan, T., Haydon, P.G.,
disease β-secretase (BACE1) is stimulus-dependent. Glia 41,
Carmignoto, G., 2004. Neuronal synchrony mediated by
astrocytic glutamate through activation of extrasynaptic
Haydon, P.G., Carmignoto, G., 2006. Astrocyte control of synaptic
NMDA receptors. Neuron 43, 729–743.
transmission and neurovascular coupling. Physiol. Rev. 86,
Fellin, T., Gomez-Gonzalo, M., Gobbo, S., Carmignoto, G., Haydon,
P.G., 2006. Astrocytic glutamate is not necessary for the
Hazell, A.S., 2009. Astrocytes are a major target in thiamine
generation of epileptiform neuronal activity in hippocampal
deficiency and Wernicke's encephalopathy. Neurochem. Int.
slices. J. Neurosci. 26, 9312–9322.
55, 129–135.
Fetler, L., Amigorena, S., 2005. Neuroscience. Brain under
Hazell, A.S., Sheedy, D., Oanea, R., Aghourian, M., Sun, S., Jung, J.Y.,
surveillance: the microglia patrol. Science 309, 392–393.
Wang, D., Wang, C., 2009. Loss of astrocytic glutamate
Floden, A.M., Combs, C.K., 2006. β-Amyloid stimulates murine
transporters in Wernicke encephalopathy. Glia 58, 148–156.
postnatal and adult microglia cultures in a unique manner.
Heneka, M.T., Feinstein, D.L., 2001. Expression and function of
J. Neurosci. 26, 4644–4648.
inducible nitric oxide synthase in neurons. J. Neuroimmunol.
Forman, M.S., Lal, D., Zhang, B., Dabir, D.V., Swanson, E., Lee, V.M.,
114, 8–18.
Trojanowski, J.Q., 2005. Transgenic mouse model of tau
Heneka, M.T., O'Banion, M.K., 2007. Inflammatory processes in
pathology in astrocytes leading to nervous system
Alzheimer's disease. J. Neuroimmunol. 184, 69–91.
degeneration. J. Neurosci. 25, 3539–3550.
Heneka, M.T., Wiesinger, H., Dumitrescu-Ozimek, L., Riederer, P.,
Frautschy, S.A., Yang, F., Irrizarry, M., Hyman, B., Saido, T.C.,
Feinstein, D.L., Klockgether, T., 2001. Neuronal and glial
Hsiao, K., Cole, G.M., 1998. Microglial response to amyloid
coexpression of argininosuccinate synthetase and inducible
plaques in APPsw transgenic mice. Am. J. Pathol. 152, 307–317.
nitric oxide synthase in Alzheimer disease. J. Neuropathol. Exp.
Frei, K., Nohava, K., Malipiero, U.V., Schwerdel, C., Fontana, A.,
Neurol. 60, 906–916.
1992. Production of macrophage colony-stimulating factor by
Heneka, M.T., Sastre, M., Dumitrescu-Ozimek, L., Dewachter, I.,
astrocytes and brain macrophages. J. Neuroimmunol. 40,
Walter, J., Klockgether, T., Van Leuven, F., 2005a. Focal glial
activation coincides with increased BACE1 activation and
Gadea, A., Lopez-Colome, A.M., 2001. Glial transporters for
precedes amyloid plaque deposition in APP[V717I] transgenic
glutamate, glycine and GABA I. Glutamate transporters. J.
mice. J. Neuroinflam. 2, 22.
Neurosci. Res. 63, 453–460.
Heneka, M.T., Sastre, M., Dumitrescu-Ozimek, L., Hanke, A.,
Giaume, C., Kirchhoff, F., Matute, C., Reichenbach, A., Verkhratsky,
Dewachter, I., Kuiperi, C., O'Banion, K., Klockgether, T., Van
A., 2007. Glia: the fulcrum of brain diseases. Cell Death Differ.
Leuven, F., Landreth, G.E., 2005b. Acute treatment with the
14, 1324–1335.
PPARgamma agonist pioglitazone and ibuprofen reduces glial
Giaume, C., Maravall, M., Welker, E., Bonvento, G., 2009. The barrel
inflammation and Aβ1–42 levels in APPV717I transgenic mice.
cortex as a model to study dynamic neuroglial interaction.
Brain 128, 1442–1453.
Neuroscientist 15, 351–366.
Hensley, K., Floyd, R.A., Gordon, B., Mou, S., Pye, Q.N., Stewart, C.,
Godoy, M.C.P., Tarelli, R., Ferrari, C.C., Sarchi, M.I.S., Pitossi, F.J.,
West, M., Williamson, K., 2002. Temporal patterns of cytokine
2008. Central and systemic IL-1 exacerbates
and apoptosis-related gene expression in spinal cords of the
neurodegeneration and motor symptoms in a model of
G93A-SOD1 mouse model of amyotrophic lateral sclerosis.
Parkinson's disease. Brain 131, 1880–1894.
J. Neurochem. 82, 365–374.
Hensley, K., Fedynyshyn, J., Ferrell, S., Floyd, R.A., Gordon, B.,
Kersaitis, C., Halliday, G.M., Kril, J.J., 2004. Regional and cellular
Grammas, P., Hamdheydari, L., Mhatre, M., Mou, S., Pye, Q.N.,
pathology in frontotemporal dementia: relationship to stage
Stewart, C., West, M., West, S., Williamson, K.S., 2003. Message
of disease in cases with and without Pick bodies. Acta
and protein-level elevation of tumor necrosis factor α (TNF α)
Neuropathol. 108, 515–523.
and TNF α-modulating cytokines in spinal cords of the
Kershman, J., 1939. Genesis of microglia in the human brain. Arch.
G93A-SOD1 mouse model for amyotrophic lateral sclerosis.
Neurol. Psychiatr. 41, 24–50.
Neurobiol. Dis. 14, 74–80.
Kettenmann, H., Ransom, B.R. (Eds.), 2005. Neuroglia. Oxford, OUP.
Heyser, C.J., Masliah, E., Samimi, A., Campbell, I.L., Gold, L.H., 1997.
Kettenmann, H., Verkhratsky, A., 2008. Neuroglia: the 150 years
Progressive decline in avoidance learning paralleled by
after. Trends Neurosci. 31, 653–659.
inflammatory neurodegeneration in transgenic mice
Kiaei, M., Kipiani, K., Chen, J.Y., Calingasan, N.Y., Beal, M.F., 2005.
expressing interleukin 6 in the brain. Proc. Natl. Acad. Sci.
Peroxisome proliferator-activated receptor-gamma agonist
U. S. A. 94, 1500–1505.
extends survival in transgenic mouse model of amyotrophic
Horuk, R., Martin, A.W., Wang, Z.X., Schweitzer, L., Gerassimides,
lateral sclerosis. Exp. Neurol. 191, 331–336.
A., Guo, H.H., Lu, Z.H., Hesselgesser, J., Perez, H.D., Kim, J.,
Kim, S., Cho, S.H., Kim, K.Y., Shin, K.Y., Kim, H.S., Park, C.H., Chang,
Parker, J., Hadley, T.J., Peiper, S.C., 1997. Expression of
K.A., Lee, S.H., Cho, D., Suh, Y.H., 2009. α-Synuclein induces
chemokine receptors by subsets of neurons in the central
migration of BV-2 microglial cells by up-regulation of CD44 and
nervous system. J. Immunol. 158, 2882–2890.
MT1-MMP 4. J. Neurochem. 109, 1483–1496.
Houades, V., Koulakoff, A., Ezan, P., Seif, I., Giaume, C., 2008. Gap
Kirischuk, S., Kettenmann, H., Verkhratsky, A., 1997. Na+/Ca2+
junction-mediated astrocytic networks in the mouse barrel
exchanger modulates kainate-triggered Ca2+ signaling in
cortex. J. Neurosci. 28, 5207–5217.
Bergmann glial cells in situ. FASEB J. 11, 566–572.
Iadecola, C., Nedergaard, M., 2007. Glial regulation of the cerebral
Kirischuk, S., Kettenmann, H., Verkhratsky, A., 2007. Membrane
microvasculature. Nat. Neurosci. 10, 1369–1376.
currents and cytoplasmic sodium transients generated by
Iribarren, P., Chen, K., Hu, J., Gong, W., Cho, E.H., Lockett, S.,
glutamate transport in Bergmann glial cells. Pflugers. Arch.
Uranchimeg, B., Wang, J.M., 2005. CpG-containing
454, 245–252.
oligodeoxynucleotide promotes microglial cell uptake of
Kofuji, P., Newman, E.A., 2004. Potassium buffering in the central
amyloid β1–42 peptide by up-regulating the expression of the
nervous system. Neuroscience 129, 1045–1056.
G-protein-coupled receptor mFPR2. FASEB J. 19, 2032–2034.
Kölliker, A., 1889. Handbuch der Gewebelehre des Menschen.
Jabs, R., Matthias, K., Grote, A., Grauer, M., Seifert, G., Steinhauser,
Leipzig, Wilhelm Engelmann.
C., 2007. Lack of P2X receptor mediated currents in astrocytes
Komori, T., 1999. Tau-positive glial inclusions in progressive
and GluR type glial cells of the hippocampal CA1 region. Glia
supranuclear palsy, corticobasal degeneration and Pick's
55, 1648–1655.
disease. Brain Pathol. 9, 663–679.
Jabs, R., Seifert, G., Steinhauser, C., 2008. Astrocytic function and
Kozlov, A.S., Angulo, M.C., Audinat, E., Charpak, S., 2006. Target
its alteration in the epileptic brain. Epilepsia 49 (Suppl. 2), 3–12.
cell-specific modulation of neuronal activity by astrocytes.
Jang, H., Boltz, D., Sturm-Ramirez, K., Shepherd, K.R., Jiao, Y.,
Proc. Natl. Acad. Sci. U. S. A. 103, 10058–10063.
Webster, R., Smeyne, R.J., 2009. Highly pathogenic H5N1
Kreutzberg, G.W., 1996. Microglia: a sensor for pathological events
influenza virus can enter the central nervous system and
in the CNS. Trends Neurosci. 19, 312–318.
induce neuroinflammation and neurodegeneration. Proc. Natl.
Kriz, J., Nguyen, M.D., Julien, J.P., 2002. Minocycline slows disease
Acad. Sci. U. S. A. 106, 14063–14068.
progression in a mouse model of arnyotrophic lateral sclerosis.
Jessen, K.R., Mirsky, R., 2008. Negative regulation of myelination:
Neurobiol. Dis. 10, 268–278.
relevance for development, injury, and demyelinating disease.
Kuchibhotla, K.V., Lattarulo, C.R., Hyman, B.T., Bacskai, B.J., 2009.
Glia 56, 1552–1565.
Synchronous hyperactivity and intercellular calcium waves in
Jiang, C., Ting, A.T., Seed, B., 1998. PPAR-γ agonists inhibit
astrocytes in Alzheimer mice. Science 323, 1211–1215.
production of monocyte inflammatory cytokines. Nature 391,
Kunst, C.B., 2004. Complex genetics of amyotrophic lateral
sclerosis. Am. J. Hum. Genet. 75, 933–947.
Johansson, A., Engler, H., Blomquist, G., Scott, B., Wall, A.,
Lalo, U., Pankratov, Y., Kirchhoff, F., North, R.A., Verkhratsky, A.,
Aquilonius, S.M., Langstrom, B., Askmark, H., 2007. Evidence
2006. NMDA receptors mediate neuron-to-glia signaling in
for astrocytosis in ALS demonstrated by [11C](L)-deprenyl-D2
mouse cortical astrocytes. J. Neurosci. 26, 2673–2683.
PET. J. Neurol. Sci. 255, 17–22.
Lalo, U., Pankratov, Y., Wichert, S.P., Rossner, M.J., North, R.A.,
Jourdain, P., Bergersen, L.H., Bhaukaurally, K., Bezzi, P., Santello,
Kirchhoff, F., Verkhratsky, A., 2008. P2X1 and P2X5 subunits
M., Domercq, M., Matute, C., Tonello, F., Gundersen, V.,
form the functional P2X receptor in mouse cortical astrocytes.
Volterra, A., 2007. Glutamate exocytosis from astrocytes
J. Neurosci. 28, 5473–5480.
controls synaptic strength. Nat. Neurosci. 10, 331–339.
Lee, E.O., Shin, Y.J., Chong, Y.H., 2004. Mechanisms involved in
Kamboh, M.I., Sanghera, D.K., Ferrell, R.E., DeKosky, S.T., 1995.
prostaglandin E2-mediated neuroprotection against TNF-α:
APOE⁎4-associated Alzheimer's disease risk is modified by
possible involvement of multiple signal transduction
α1-antichymotrypsin polymorphism. Nat. Genet. 10, 486–488.
and beta-catenin/T-cell factor. J. Neuroimmunol. 155, 21–31.
Kang, J., Kang, N., Lovatt, D., Torres, A., Zhao, Z., Lin, J.,
Lees, A.J., 2009. The Parkinson chimera. Neurology 72, S2–11.
Nedergaard, M., 2008. Connexin 43 hemichannels are
Lee, S.H., Magge, S., Spencer, D.D., Sontheimer, H., Cornell-Bell, A.H.,
permeable to ATP. J. Neurosci. 28, 4702–4711.
1995. Human epileptic astrocytes exhibit increased gap junction
Karram, K., Goebbels, S., Schwab, M., Jennissen, K., Seifert, G.,
coupling. Glia 15, 195–202.
Steinhauser, C., Nave, K.A., Trotter, J., 2008. NG2-expressing
Lee, S.Y., Haydon, P.G., 2007. Astrocytic glutamate targets NMDA
cells in the nervous system revealed by the NG2-EYFP-knockin
receptors. J. Physiol. 581, 887–888.
mouse. Genesis 46, 743–757.
Lenhossek, M.v., 1893. Der feinere Bau des Nervensystems
Kaul, M., Garden, G.A., Lipton, S.A., 2001. Pathways to neuronal
im Lichte neuester Forschung. Fischer's Medicinische
injury and apoptosis in HIV-associated dementia. Nature 410,
Buchhandlung H. Kornfield, Berlin.
Leoni, G., Rattray, M., Butt, A.M., 2009. NG2 cells differentiate into
Kelley, K.A., Ho, L., Winger, D., Freire-Moar, J., Borelli, C.B., Aisen,
astrocytes in cerebellar slices. Mol. Cell Neurosci. 42, 208–218.
P.S., Pasinetti, G.M., 1999. Potentiation of excitotoxicity in
Levine, J.M., Beasley, L., Stallcup, W.B., 1986. Localization of a
transgenic mice overexpressing neuronal cyclooxygenase-2.
neurectoderm-associated cell surface antigen in the
Am. J. Pathol. 155, 995–1004.
developing and adult rat. Brain Res. 392, 211–222.
Liang, X.B., Wang, Q., Hand, T., Wu, L.J., Breyer, R.M., Montine, T.J.,
Passmore, A.P., 2001. Association between polymorphism in
Andreasson, K., 2005. Deletion of the prostaglandin E-2 EP2
regulatory region of gene encoding tumour necrosis factor
receptor reduces oxidative damage and amyloid burden in a
alpha and risk of Alzheimer's disease and vascular dementia: a
model of Alzheimer's disease. J. Neurosci. 25, 10180–10187.
case–control study. Lancet 357, 436–439.
Liauw, J., Hoang, S., Choi, M., Eroglu, C., Sun, G.H., Percy, M.,
McGeer, E.G., McGeer, P.L., 1999. Brain inflammation in Alzheimer
Wildman-Tobriner, B., Bliss, T., Guzman, R.G., Barres, B.A.,
disease and the therapeutic implications. Curr. Pharm. Des. 5,
Steinberg, G.K., 2008. Thrombospondins 1 and 2 are necessary
for synaptic plasticity and functional recovery after stroke. J.
McGeer, P.L., McGeer, E.G., 1995. The inflammatory response
Cereb. Blood Flow. Metab. 28, 1722–1732.
system of brain: implications for therapy of Alzheimer and
Liberatore, G.T., Jackson-Lewis, V., Vukosavic, S., Mandir, A.S.,
other neurodegenerative diseases. Brain Res. Brain Res. Rev. 21,
Vila, M., McAuliffe, W.G., Dawson, V.L., Dawson, T.M.,
Przedborski, S., 1999. Inducible nitric oxide synthase stimulates
McGeer, P.L., McGeer, E.G., 2002a. Inflammatory processes in
dopaminergic neurodegeneration in the MPTP model of
amyotrophic lateral sclerosis. Muscle Nerve 26, 459–470.
Parkinson disease. Nat. Med. 5, 1403–1409.
McGeer, P.L., McGeer, E.G., 2002b. The possible role of complement
Li, L., Lundkvist, A., Andersson, D., Wilhelmsson, U., Nagai, N.,
activation in Alzheimer disease. Trends Mol. Med. 8,
Pardo, A.C., Nodin, C., Stahlberg, A., Aprico, K., Larsson, K.,
Yabe, T., Moons, L., Fotheringham, A., Davies, I., Carmeliet, P.,
McGeer, P.L., McGeer, E.G., 2008. Glial reactions in Parkinson's
Schwartz, J.P., Pekna, M., Kubista, M., Blomstrand, F., Maragakis,
disease. Mov. Disord. 23, 474–483.
N., Nilsson, M., Pekny, M., 2008. Protective role of reactive
McTigue, D.M., Tripathi, R.B., 2008. The life, death, and replacement
astrocytes in brain ischemia. J. Cereb. Blood Flow Metab. 28,
of oligodendrocytes in the adult CNS. J. Neurochem. 107, 1–19.
Meda, L., Baron, P., Prat, E., Scarpini, E., Scarlato, G., Cassatella, M.A.,
Lindberg, C., Selenica, M.L.B., Westlind-Danielsson, A., Schultzberg,
Rossi, F., 1999. Proinflammatory profile of cytokine
M., 2005. β-Amyloid protein structure determines the nature
production by human monocytes and murine microglia stimu-
of cytokine release from rat microglia. J. Mol. Neurosci. 27,
lated with β-amyloid25–35. J. Neuroimmunol. 93, 45–52.
Mena, M.A., de Yebenes, J.G., 2006. Drug-induced parkinsonism.
Lino, M.M., Schneider, C., Caroni, P., 2002. Accumulation of SOD1
Expert. Opin. Drug Saf. 5, 759–771.
mutants in postnatal motoneurons does not cause motoneuron
Mena, M.A., Garcia de Yebenes, J., 2008. Glial cells as players in
pathology or motoneuron disease. J. Neurosci. 22, 4825–4832.
parkinsonism: the "good," the "bad," and the "mysterious" glia.
Lin, S.C., Bergles, D.E., 2002. Physiological characteristics of
Neuroscientist 14, 544–560.
NG2-expressing glial cells. J. Neurocytol. 31, 537–549.
Mena, M.A., Casarejos, M.J., Carazo, A., Paino, C.L., Garcia de
Liu, B., Hong, J.S., 2003. Role of microglia in inflammation-mediated
Yebenes, J., 1996. Glia conditioned medium protects fetal rat
neurodegenerative diseases: mechanisms and strategies
midbrain neurones in culture from L-DOPA toxicity.
for therapeutic intervention. J. Pharmacol. Exp. Ther. 304, 1–7.
Neuroreport 7, 441–445.
Lotz, M., Ebert, S., Esselmann, H., Iliev, A.I., Prinz, M., Wiazewicz,
Mena, M.A., Casarejos, M.J., Garcia de Yebenes, J., 1999. The effect
N., Wiltfang, J., Gerber, J., Nau, R., 2005. Amyloid β peptide 1–40
of glia-conditioned medium on dopamine neurons in culture.
enhances the action of Toll-like receptor-2 and -4 agonists but
Modulation of apoptosis, tyrosine hydroxylase expression and
antagonizes Toll-like receptor-9-induced inflammation in
1-methyl-4-phenylpyridinium toxicity. J. Neural. Transm. 106,
primary mouse microglial cell cultures. J. Neurochem. 94,
Mena, M.A., de Bernardo, S., Casarejos, M.J., Canals, S.,
Lue, L.F., Rydel, R., Brigham, E.F., Yang, L.B., Hampel, H., Murphy Jr.,
Rodriguez-Martin, E., 2002. The role of astroglia on the survival
G.M., Brachova, L., Yan, S.D., Walker, D.G., Shen, Y., Rogers, J.,
of dopamine neurons. Mol. Neurobiol. 25, 245–263.
2001. Inflammatory repertoire of Alzheimer's disease and
Metea, M.R., Newman, E.A., 2006. Glial cells dilate and constrict
nondemented elderly microglia in vitro. Glia 35, 72–79.
blood vessels: a mechanism of neurovascular coupling.
Magistretti, P.J., 2006. Neuron–glia metabolic coupling and
J. Neurosci. 26, 2862–2870.
plasticity. J. Exp. Biol. 209, 2304–2311.
Meyer-Luehmann, M., Spires-Jones, T.L., Prada, C., Garcia-Alloza,
Magistretti, P.J., 2009. Role of glutamate in neuron–glia metabolic
M., de Calignon, A., Rozkalne, A., Koenigsknecht-Talboo, J.,
coupling. Am. J. Clin. Nutr. 90, 875S–880S.
Holtzman, D.M., Bacskai, B.J., Hyman, B.T., 2008. Rapid
Manning Jr., T.J., Sontheimer, H., 1997. Spontaneous intracellular
appearance and local toxicity of amyloid-beta plaques in a
calcium oscillations in cortical astrocytes from a patient with
mouse model of Alzheimer's disease. Nature 451, 720–724.
intractable childhood epilepsy (Rasmussen's encephalitis). Glia
Minghetti, L., Polazzi, E., Nicolini, A., Creminon, C., Levi, G., 1997.
21, 332–337.
Up-regulation of cyclooxygenase-2 expression in cultured
Martinez-Hernandez, A., Bell, K.P., Norenberg, M.D., 1977. Glutamine
microglia by prostaglandin E2, cyclic AMP and non-steroidal
synthetase: glial localization in brain. Science 195, 1356–1358.
anti-inflammatory drugs. Eur. J. Neurosci. 9, 934–940.
Matos, M., Augusto, E., Oliveira, C.R., Agostinho, P., 2008.
Mrak, R.E., Griffin, W.S., 2001. Interleukin-1, neuroinflammation,
Amyloid-beta peptide decreases glutamate uptake in
and Alzheimer's disease. Neurobiol. Aging 22, 903–908.
cultured astrocytes: involvement of oxidative stress and
Mulligan, S.J., MacVicar, B.A., 2004. Calcium transients in astrocyte
mitogen-activated protein kinase cascades. Neuroscience 156,
endfeet cause cerebrovascular constrictions. Nature 431,
Matute, C., 2008. P2X7 receptors in oligodendrocytes: a novel target
Nagai, M., Re, D.B., Nagata, T., Chalazonitis, A., Jessell, T.M.,
for neuroprotection. Mol. Neurobiol. 38, 123–128.
Wichterle, H., Przedborski, S., 2007. Astrocytes expressing
Matute, C., Alberdi, E., Domercq, M., Sanchez-Gomez, M.V.,
ALS-linked mutated SOD1 release factors selectively toxic to
Perez-Samartin, A., Rodriguez-Antiguedad, A., Perez-Cerda, F.,
motor neurons. Nat. Neurosci. 10, 615–622.
2007. Excitotoxic damage to white matter. J. Anat. 210, 693–702.
Nagatsu, T., Sawada, M., 2005. Inflammatory process in
McCullough, L., Wu, L., Haughey, N., Liang, X., Hand, T., Wang, Q.,
Parkinson's disease: role for cytokines. Curr. Pharm. Design 11,
Breyer, R.M., Andreasson, K., 2004. Neuroprotective function of
the PGE2 EP2 receptor in cerebral ischemia. J. Neurosci. 24,
Nagele, R.G., D'Andrea, M.R., Lee, H., Venkataraman, V., Wang,
H.Y., 2003. Astrocytes accumulate A β 42 and give rise to
McCusker, S.M., Curran, M.D., Dynan, K.B., McCullagh, C.D.,
astrocytic amyloid plaques in Alzheimer disease brains. Brain
Urquhart, D.D., Middleton, D., Patterson, C.C., McIlroy, S.P.,
Res. 971, 197–209.
Nagele, R.G., Wegiel, J., Venkataraman, V., Imaki, H., Wang, K.C.,
Panatier, A., Theodosis, D.T., Mothet, J.P., Touquet, B., Pollegioni,
2004. Contribution of glial cells to the development of amyloid
L., Poulain, D.A., Oliet, S.H., 2006. Glia-derived D-serine controls
plaques in Alzheimer's disease. Neurobiol. Aging 25, 663–674.
NMDA receptor activity and synaptic memory. Cell 125,
Nardacci, R., Antinori, A., Kroemer, G., Piacentini, M., 2005. Cell
death mechanisms in HIV-associated dementia: the
Pankratov, Y., Lalo, U., Verkhratsky, A., North, R.A., 2006.
involvement of syncytia. Cell Death Differ. 12 (Suppl. 1),
Vesicular release of ATP at central synapses. Pflugers Arch.
452, 589–597.
Narumiya, S., Sugimoto, Y., Ushikubi, F., 1999. Prostanoid
Papassotiropoulos, A., Bagli, M., Jessen, F., Bayer, T.A., Maier, W., Rao,
receptors: structures, properties, and functions. Physiol. Rev.
M.L., Heun, R., 1999. A genetic variation of the inflammatory
79, 1193–1226.
cytokine interleukin-6 delays the initial onset and reduces the
Nave, K.A., Sereda, M.W., Ehrenreich, H., 2007. Mechanisms of
risk for sporadic Alzheimer's disease. Ann. Neurol. 45, 666–668.
disease: inherited demyelinating neuropathies—from basic to
Parkinson, J., 1817. An Essay on the Shaking Palsy. Sherwood,
clinical research. Nat. Clin. Pract. Neurol. 3, 453–464.
Neely, and Jones, London, p. 66.
Ndubaku, U., de Bellard, M.E., 2008. Glial cells: old cells with new
Parkinson, J., 2002. An essay on the shaking palsy. 1817.
twists. Acta Histochem. 110, 182–195.
J. Neuropsychiatry Clin. Neurosci. 14, 223–236.
Nedergaard, M., Dirnagl, U., 2005. Role of glial cells in cerebral
Parpura, V., Haydon, P.G., 2000. Physiological astrocytic calcium
ischemia. Glia 50, 281–286.
levels stimulate glutamate release to modulate adjacent
Nedergaard, M., Ransom, B., Goldman, S.A., 2003. New roles for
neurons. Proc. Natl. Acad. Sci. U. S. A. 97, 8629–8634.
astrocytes: redefining the functional architecture of the brain.
Pascual, O., Casper, K.B., Kubera, C., Zhang, J., Revilla-Sanchez, R.,
Trends Neurosci. 26, 523–530.
Sul, J.Y., Takano, H., Moss, S.J., McCarthy, K., Haydon, P.G., 2005.
Newman, E.A., 1995. Glial cell regulation of extracellular potassium.
Astrocytic purinergic signaling coordinates synaptic networks.
In: Kettenmann, H., Ransom, B. (Eds.), Neuroglia. Oxford
Science 310, 113–116.
University Press, NY, pp. 717–731.
Pekny, M., Nilsson, M., 2005. Astrocyte activation and reactive
Newman, E.A., Frambach, D.A., Odette, L.L., 1984. Control of
gliosis. Glia 50, 427–434.
extracellular potassium levels by retinal glial cell K+ siphoning.
Pellerin, L., Bouzier-Sore, A.K., Aubert, A., Serres, S., Merle, M.,
Science 225, 1174–1175.
Costalat, R., Magistretti, P.J., 2007. Activity-dependent regulation
Nicoll, J.A., Mrak, R.E., Graham, D.I., Stewart, J., Wilcock, G.,
of energy metabolism by astrocytes: an update. Glia 55,
MacGowan, S., Esiri, M.M., Murray, L.S., Dewar, D., Love, S.,
Moss, T., Griffin, W.S., 2000. Association of interleukin-1 gene
Perea, G., Navarrete, M., Araque, A., 2009. Tripartite synapses:
polymorphisms with Alzheimer's disease. Ann. Neurol. 47,
astrocytes process and control synaptic information. Trends
Neurosci. 32, 421–431.
Nieweg, K., Schaller, H., Pfrieger, F.W., 2009. Marked differences in
Perry, R.T., Collins, J.S., Wiener, H., Acton, R., Go, R.C., 2001. The
cholesterol synthesis between neurons and glial cells from
role of TNF and its receptors in Alzheimer's disease. Neurobiol.
postnatal rats. J. Neurochem. 109, 125–134.
Aging 22, 873–883.
Niino, M., Iwabuchi, K., Kikuchi, S., Ato, M., Morohashi, T., Ogata,
Pfrieger, F.W., 2009. Roles of glial cells in synapse development.
A., Tashiro, K., Onoe, K., 2001. Amelioration of experimental
Cell Mol. Life Sci. 66, 2037–2047.
autoimmune encephalomyelitis in C57BL/6 mice by an agonist
Pfrieger, F.W., Barres, B.A., 1996. New views on synapse–glia
of peroxisome proliferator-activated receptor-gamma.
interactions. Curr. Opin. Neurobiol. 6, 615–621.
J. Neuroimmunol. 116, 40–48.
Phul, R.K., Shaw, P.J., Ince, P.G., Smith, M.E., 2000. Expression of
Nimmerjahn, A., Kirchhoff, F., Helmchen, F., 2005. Resting
nitric oxide synthase isoforms in spinal cord in amyotrophic
microglial cells are highly dynamic surveillants of brain
lateral sclerosis. Amyotroph. Lateral Scler. Other Motor Neuron
parenchyma in vivo. Science 308, 1314–1318.
Disord. 1, 259–267.
Nishiyama, A., Komitova, M., Suzuki, R., Zhu, X., 2009.
Polazzi, E., Altamira, L.E.P., Eleuteri, S., Barbaro, R., Casadio, C.,
Polydendrocytes (NG2 cells): multifunctional cells with lineage
Contestabile, A., Monti, B., 2009. Neuroprotection of microglial
plasticity. Nat. Rev. Neurosci. 10, 9–22.
conditioned medium on 6-hydroxydopamine-induced
O'Banion, M.K., Miller, J.C., Chang, J.W., Kaplan, M.D., Coleman,
neuronal death: role of transforming growth factor β-2.
P.D., 1996. Interleukin-1 β induces prostaglandin G/H synthase-2
J. Neurochem. 110, 545–556.
(cyclooxygenase-2) in primary murine astrocyte cultures.
Pompl, P.N., Ho, L., Bianchi, M., McManus, T., Qin, W., Pasinetti,
J. Neurochem. 66, 2532–2540.
G.M., 2003. A therapeutic role for cyclooxygenase-2 inhibitors
Oberheim, N.A., Wang, X., Goldman, S., Nedergaard, M., 2006.
in a transgenic mouse model of amyotrophic lateral sclerosis.
Astrocytic complexity distinguishes the human brain. Trends
FASEB J. 17, 725–727.
Neurosci. 29, 547–553.
Potts, R., Leech, R.W., 2005. Thalamic dementia: an example of
Oberheim, N.A., Takano, T., Han, X., He, W., Lin, J.H., Wang, F., Xu,
primary astroglial dystrophy of Seitelberger. Clin. Neuropathol.
Q., Wyatt, J.D., Pilcher, W., Ojemann, J.G., Ransom, B.R.,
24, 271–275.
Goldman, S.A., Nedergaard, M., 2009. Uniquely hominid
Pramatarova, A., Laganiere, J., Roussel, J., Brisebois, K., Rouleau,
features of adult human astrocytes. J. Neurosci. 29, 3276–3287.
G.A., 2001. Neuron-specific expression of mutant superoxide
Oddo, S., Caccamo, A., Shepherd, J.D., Murphy, M.P., Golde, T.E.,
dismutase 1 in transgenic mice does not lead to motor
Kayed, R., Metherate, R., Mattson, M.P., Akbari, Y., LaFerla, F.M.,
impairment. J. Neurosci. 21, 3369–3374.
2003. Triple-transgenic model of Alzheimer's disease with
Qiu, W.Q., Walsh, D.M., Ye, Z., Vekrellis, K., Zhang, J., Podlisny,
plaques and tangles: intracellular Abeta and synaptic
M.B., Rosner, M.R., Safavi, A., Hersh, L.B., Selkoe, D.J., 1998.
dysfunction. Neuron 39, 409–421.
Insulin-degrading enzyme regulates extracellular levels of
Okun, E., Griffioen, K.J., Lathia, J.D., Tang, S.C., Mattson, M.P.,
amyloid beta-protein by degradation. J. Biol. Chem. 273,
Arumugam, T.V., 2009. Toll-like receptors in neurodegeneration.
Brain Res. Rev. 59, 278–292.
Rajkowska, G., Miguel-Hidalgo, J.J., Wei, J., Dilley, G., Pittman, S.D.,
Oliet, S.H., Mothet, J.P., 2009. Regulation of N-methyl-D-aspartate
Meltzer, H.Y., Overholser, J.C., Roth, B.L., Stockmeier, C.A., 1999.
receptors by astrocytic D-serine. Neuroscience 158, 275–283.
Morphometric evidence for neuronal and glial prefrontal
Owe, S.G., Marcaggi, P., Attwell, D., 2006. The ionic stoichiometry
cell pathology in major depression. Biol. Psychiatry 45,
of the GLAST glutamate transporter in salamander retinal glia.
J. Physiol. 577, 591–599.
Ransohoff, R.M., Perry, V.H., 2009. Microglial physiology: unique
stimuli, specialized responses. Annu. Rev. Immunol. 27,
Scemes, E., Giaume, C., 2006. Astrocyte calcium waves: what they
are and what they do. Glia 54, 716–725.
Ransom, B., Behar, T., Nedergaard, M., 2003. New roles for
Schleich, C.L., 1894. Schmerzlose Operationen: Örtliche Betäubung
astrocytes (stars at last). Trends Neurosci. 26, 520–522.
mit indiffrenten Flüssigkeiten. Psychophysik des natürlichen
Retzius, G., 1894–1916. Biol Untersuchungen. Die neuroglia des
und künstlichen Schlafes. Julius Springer, Berlin, p. 256.
Gehirns beim Menschen und bei Saeugethieren, pp. 1–28.
Schubert, P., Morino, T., Miyazaki, H., Ogata, T., Nakamura, Y.,
Rio-Hortega, P.d., 1919a. El tercer elemento de los centros
Marchini, C., Ferroni, S., 2000. Cascading glia reactions: a
nerviosos. I. La microglia en estado normal. II. Intervencíon de
common pathomechanism and its differentiated control by
la microglia en los procesos patológicos. III. Naturaleza
cyclic nucleotide signaling. Ann. N.Y. Acad. Sci. 903, 24–33.
probable de la microglia. Bol de la Soc esp de biol 9, 69–120.
Schutz, B., Reimann, J., Dumitrescu-Ozimek, L., Kappes-Horn, K.,
Rio-Hortega, P.d., 1919b. Poder fagocitario y movilidad de la
Landreth, G.E., Schurmann, B., Zimmer, A., Heneka, M.T., 2005.
microglia. Bol de la Soc esp de biol 9, 154.
The oral antidiabetic pioglitazone protects from
Rio-Hortega, P.d., 1920. La microglia y su transformacíon en
neurodegeneration and amyotrophic lateral sclerosis-like
células en bastoncito y cuerpos gránulo-adiposos. Trab del Lab
symptoms in superoxide dismutase-G93A transgenic mice.
de invest biol 18, 37.
J. Neurosci. 25, 7805–7812.
Rio-Hortega, P.d., 1921. Estudios sobre la neuroglia. La glia de
Seifert, G., Schilling, K., Steinhauser, C., 2006. Astrocyte
escasa radiaciones oligodendroglia. Bol, Soc, Esp, Biol. 21,
dysfunction in neurological disorders: a molecular perspective.
Nat. Rev. Neurosci. 7, 194–206.
Rodriguez, J.J., Olabarria, M., Chvatal, A., Verkhratsky, A., 2009.
Selmaj, K.W., Farooq, M., Norton, W.T., Raine, C.S., Brosnan, C.F.,
Astroglia in dementia and Alzheimer's disease. Cell Death
1990. Proliferation of astrocytes in vitro in response to
Differ 16, 378–385.
cytokines. A primary role for tumor necrosis factor. J. Immunol.
Rolls, A., Shechter, R., Schwartz, M., 2009. The bright side of the
144, 129–135.
glial scar in CNS repair. Nat. Rev. Neurosci. 10, 235–241.
Shie, F.S., Breyer, R.M., Montine, T.J., 2005a. Microglia lacking E
Rosen, D.R., Siddique, T., Patterson, D., Figlewicz, D.A., Sapp, P.,
Prostanoid Receptor subtype 2 have enhanced Aβ phagocytosis
Hentati, A., Donaldson, D., Goto, J., O'Regan, J.P., Deng, H.X., et al.,
yet lack Aβ-activated neurotoxicity. Am. J. Pathol. 166,
1993. Mutations in Cu/Zn superoxide dismutase gene are
associated with familial amyotrophic lateral sclerosis. Nature
Shie, F.S., Montine, K.S., Breyer, R.M., Montine, T.J., 2005b.
362, 59–62.
Microglial EP2 is critical to neurotoxicity from activated
Rossi, D., Volterra, A., 2009. Astrocytic dysfunction: insights on the
cerebral innate immunity. Glia 52, 70–77.
role in neurodegeneration. Brain Res. Bull. 80, 224–232.
Simard, M., Nedergaard, M., 2004. The neurobiology of glia in the
Rossi, D., Brambilla, L., Valori, C.F., Roncoroni, C., Crugnola, A.,
context of water and ion homeostasis. Neuroscience 129,
Yokota, T., Bredesen, D.E., Volterra, A., 2008. Focal degeneration
of astrocytes in amyotrophic lateral sclerosis. Cell Death Differ.
Slawik, H., Volk, B., Fiebich, B., Hull, M., 2004. Microglial expression
15, 1691–1700.
of prostaglandin EP3 receptor in excitotoxic lesions in the rat
Rossi, D.J., Oshima, T., Attwell, D., 2000. Glutamate release in
striatum. Neurochem. Int. 45, 653–660.
severe brain ischaemia is mainly by reversed uptake. Nature
Smith, K.J., Hall, S.M., 2001. Factors directly affecting impulse
403, 316–321.
transmission in inflammatory demyelinating disease: recent
Rossi, F., Bianchini, E., 1996. Synergistic induction of nitric oxide by
advances in our understanding. Curr. Opin. Neurol. 14, 289–298.
β-amyloid and cytokines in astrocytes. Biochem. Biophys. Res.
Stallcup, W.B., 2002. The NG2 proteoglycan: past insights and
Commun. 225, 474–478.
future prospects. J. Neurocytol. 31, 423–435.
Rossner, S., Lange-Dohna, C., Zeitschel, U., Perez-Polo, J.R., 2005.
Standaert, D.G., Theodore, S., Cao, S.W., McLean, P., 2009.
Alzheimer's disease β-secretase BACE1 is not a neuron-specific
α-Synuclein triggers microglial activation and adaptive
enzyme. J. Neurochem. 92, 226–234.
immunity in a mouse model of Parkinson disease. Neurology
Rota, E., Bellone, G., Rocca, P., Bergamasco, B., Emanuelli, G.,
Ferrero, P., 2006. Increased intrathecal TGF-β 1, but not IL-12,
Swanson, R.A., 2005. Astrocyte neurotransmitter uptake. In:
IFN-γ and IL-10 levels in Alzheimer's disease patients. Neurol.
Kettenmann, H., Ransom, B. (Eds.), Neuroglia. Oxford
Sciences 27, 33–39.
University Press, Oxford, pp. 346–354.
Rouach, N., Koulakoff, A., Abudara, V., Willecke, K., Giaume, C.,
Szczepanik, A.M., Rampe, D., Ringheim, G.E., 2001. Amyloid-β
2008. Astroglial metabolic networks sustain hippocampal
peptide fragments p3 and p4 induce pro-inflammatory
synaptic transmission. Science 322, 1551–1555.
cytokine and chemokine production in vitro and in vivo.
Roy, C.S., Sherrington, C.S., 1890. On the regulation of the
J. Neurochem. 77, 304–317.
blood-supply of the brain. J. Physiol. (Lond) 11, 85–108.
Tahara, K., Kim, H.D., Jin, J.J., Maxwell, J.A., Li, L., Fukuchi, K., 2006.
Sanz, J.M., Chiozzi, P., Ferrari, D., Colaianna, M., Idzko, M., Falzoni,
Role of toll-like receptor signalling in Abeta uptake and
S., Fellin, R., Trabace, L., Di Virgilio, F., 2009. Activation of
clearance. Brain 129, 3006–3019.
microglia by amyloid β requires P2X7 receptor expression.
Takadera, T., Shiraishi, Y., Ohyashiki, T., 2004. Prostaglandin E2
J. Immunol. 182, 4378–4385.
induced caspase-dependent apoptosis possibly through
Sastre, M., Dewachter, I., Landreth, G.E., Willson, T.M.,
activation of EP2 receptors in cultured hippocampal neurons.
Klockgether, T., Van Leuven, F., Heneka, M.T., 2003.
Neurochem. Int. 45, 713–719.
Nonsteroidal anti-inflammatory drugs and peroxisome
Takano, T., Tian, G.F., Peng, W., Lou, N., Libionka, W., Han, X.,
proliferator-activated receptor-gamma agonists modulate
Nedergaard, M., 2006. Astrocyte-mediated control of cerebral
immunostimulated processing of amyloid precursor protein
blood flow. Nat. Neurosci. 9, 260–267.
through regulation of β-secretase. J. Neurosci. 23,
Talbot, K., 2002. Motor neurone disease. Postgrad. Med. J. 78,
Sastre, M., Dewachter, I., Rossner, S., Bogdanovic, N., Rosen, E.,
Tanaka, K., Watase, K., Manabe, T., Yamada, K., Watanabe, M.,
Borghgraef, P., Evert, B.O., Dumitrescu-Ozimek, L., Thal, D.R.,
Takahashi, K., Iwama, H., Nishikawa, T., Ichihara, N., Kikuchi,
Landreth, G., Walter, J., Klockgether, T., Van Leuven, F., Heneka,
T., Okuyama, S., Kawashima, N., Hori, S., Takimoto, M., Wada,
M.T., 2006. Nonsteroidal anti-inflammatory drugs repress
K., 1997. Epilepsy and exacerbation of brain injury in mice
β-secretase gene promoter activity by the activation of PPAR γ.
lacking the glutamate transporter GLT-1. Science 276,
Proc. Natl. Acad. Sci. U. S. A. 103, 443–448.
Tanzi, R.E., Bertram, L., 2005. Twenty years of the Alzheimer's
Verkhratsky, A., Steinhauser, C., 2000. Ion channels in glial cells.
disease amyloid hypothesis: a genetic perspective. Cell 120,
Brain Res. Brain Res. Rev. 32, 380–412.
Verkhratsky, A., Toescu, E.C., 2006. Neuronal–glial networks as
Taylor, P.R., Carugati, A., Fadok, V.A., Cook, H.T., Andrews, M.,
substrate for CNS integration. J. Cell. Mol. Med. 10, 826–836.
Carroll, M.C., Savill, J.S., Henson, P.M., Botto, M., Walport, M.J.,
Verkhratsky, A., Orkand, R.K., Kettenmann, H., 1998. Glial calcium:
2000. A hierarchical role for classical pathway complement
homeostasis and signaling function. Physiol. Rev. 78, 99–141.
proteins in the clearance of apoptotic cells in vivo. J. Exp. Med.
Verkhratsky, A., Krishtal, O.A., Burnstock, G., 2009. Purinoceptors
192, 359–366.
on neuroglia. Mol. Neurobiol. 39, 190–208.
Theodore, S., Cao, S.W., McLean, P., Standaert, D., 2008. Microglial
Virchow, R., 1856. Gesammelte Abhandlungen zyr
activation and adaptive immune response in a mouse model of
wissenschaftlischen Medizin. Verlag von Meidinger Sohn &
Parkinson's disease. J. Neuroimmunol. 203, 188–189.
Comp, Frankfurt.
Thery, C., Stanley, E.R., Mallat, M., 1992. Interleukin-1 and
Virchow, R., 1858. Die Cellularpathologie in ihrer Begründung auf
tumor-necrosis-factor-α stimulate the production of
physiologische and pathologische Gewebelehre. Zwanzig
colony-stimulating factor-I by murine astrocytes. J. Neurochem.
Vorlesungen gehalten während der Monate Februar, März und
59, 1183–1186.
April 1858 im pathologischen Institut zu Berlin. August
Thompson, K.A., McArthur, J.C., Wesselingh, S.L., 2001. Correlation
Hirschwald, Berlin, p. 440.
between neurological progression and astrocyte apoptosis in
Volterra, A., Meldolesi, J., 2005. Astrocytes, from brain glue to
HIV-associated dementia. Ann. Neurol. 49, 745–752.
communication elements: the revolution continues. Nat. Rev.
Tian, G.F., Azmi, H., Takano, T., Xu, Q., Peng, W., Lin, J., Oberheim,
Neurosci. 6, 626–640.
N., Lou, N., Wang, X., Zielke, H.R., Kang, J., Nedergaard, M., 2005.
Walter, S., Letiembre, M., Liu, Y., Heine, H., Penke, B., Hao, W., Bode,
An astrocytic basis of epilepsy. Nat. Med. 11, 973–981.
B., Manietta, N., Walter, J., Schulz-Schuffer, W., Fassbender, K.,
Toggas, S.M., Masliah, E., Mucke, L., 1996. Prevention of HIV-1
2007. Role of the toll-like receptor 4 in neuroinflammation in
gp120-induced neuronal damage in the central nervous system
Alzheimer's disease. Cell. Physiol. Biochem. 20, 947–956.
of transgenic mice by the NMDA receptor antagonist memantine.
Warr, O., Takahashi, M., Attwell, D., 1999. Modulation of
Brain. Res. 706, 303–307.
extracellular glutamate concentration in rat brain slices by
Tsai, G., Coyle, J.T., 2002. Glutamatergic mechanisms in
cystine-glutamate exchange. J. Physiol. 514, 783–793.
schizophrenia. Annu. Rev. Pharmacol. Toxicol. 42, 165–179.
Webster, S., Lue, L.F., Brachova, L., Tenner, A.J., McGeer, P.L., Terai,
Tsai, G., Yang, P., Chung, L.C., Lange, N., Coyle, J.T., 1998. D-Serine
K., Walker, D.G., Bradt, B., Cooper, N.R., Rogers, J., 1997.
added to antipsychotics for the treatment of schizophrenia.
Molecular and cellular characterization of the membrane
Biol. Psychiatry 44, 1081–1089.
attack complex, C5b-9, in Alzheimer's disease. Neurobiol.
Tuppo, E.E., Arias, H.R., 2005. The role of inflammation in
Aging 18, 415–421.
Alzheimer's disease. Int. J. Biochem. Cell. Biol. 37, 289–305.
Weigert, K., 1895. Beiträge zur Kenntnis der normalen
Turner, B.J., Talbot, K., 2008. Transgenics, toxicity and therapeutics
menschlichen Neuroglia. Frankfurt, Diesterweg.
in rodent models of mutant SOD1-mediated familial ALS. Prog.
Wernicke, C., 1881–1883. Lehrbuch der Gehirnkrankheiten für
Neurobiol. 85, 94–134.
Aerzte und Studirende. Fischer, Kassel und Berlin.
Turner, M.R., Cagnin, A., Turkheimer, F.E., Miller, C.C.J., Shaw, C.E.,
Wilhelmsson, U., Bushong, E.A., Price, D.L., Smarr, B.L., Phung, V.,
2004. Evidence of widespread cerebral microglial activation in
Terada, M., Ellisman, M.H., Pekny, M., 2006. Redefining the
amyotrophic lateral sclerosis: an [C-11](R)-PK11195 positron
concept of reactive astrocytes as cells that remain within their
emission tomography study 1. Neurobiol. Dis. 15, 601–609.
unique domains upon reaction to injury. Proc. Natl. Acad. Sci.
van Beek, J., Elward, K., Gasque, P., 2003. Activation of complement
U. S. A. 103, 17513–17518.
in the central nervous system: roles in neurodegeneration and
Wyss-Coray, T., Mucke, L., 2002. Inflammation in neurodegenerative
neuroprotection. Ann. N.Y. Acad. Sci. 992, 56–71.
disease—a double-edged sword. Neuron 35, 419–432.
van der Valk, P., Amor, S., 2009. Preactive lesions in multiple
Wyss-Coray, T., Lin, C., Sanan, D.A., Mucke, L., Masliah, E., 2000.
sclerosis. Curr. Opin. Neurol. 22, 207–213.
Chronic overproduction of transforming growth factor-β 1 by
van Swieten, J.C., Caplan, L.R., 1993. Binswanger's disease. Adv.
astrocytes promotes Alzheimer's disease-like microvascular
Neurol. 62, 193–211.
degeneration in transgenic mice. Am. J. Pathol. 156, 139–150.
Vanzani, M.C., Iacono, R.F., Caccuri, R.L., Troncoso, A.R., Berria,
Wyss-Coray, T., Loike, J.D., Brionne, T.C., Lu, E., Anankov, R., Yan,
M.I., 2006. Regional differences in astrocyte activation in
F., Silverstein, S.C., Husemann, J., 2003. Adult mouse astrocytes
HIV-associated dementia. Medicina (B Aires) 66, 108–112.
degrade amyloid-β in vitro and in situ. Nat. Med. 9, 453–457.
Veerhuis, R., Boshuizen, R.S., Morbin, M., Mazzoleni, G., Hoozemans,
Xia, M.Q., Hyman, B.T., 1999. Chemokines/chemokine receptors in
J.J.M., Langedijk, J.P.M., Tagliavini, F., Langeveld, J.P.M.,
the central nervous system and Alzheimer's disease. J.
Eikelenboom, P., 2005. Activation of human microglia by fibrillar
Neurovirol. 5, 32–41.
prion protein-related peptides is enhanced by amyloid-associated
Xia, M.Q., Qin, S.X., Wu, L.J., Mackay, C.R., Hyman, B.T., 1998.
factors SAP and C1q. Neurobiol. Dis. 19, 273–282.
Immunohistochemical study of the β-chemokine receptors
Verkhratsky, A., 2006a. Calcium ions and integration in neural
CCR3 and CCR5 and their ligands in normal and Alzheimer's
circuits. Acta Physiol. (Oxf) 187, 357–369.
disease brains. Am. J. Pathol. 153, 31–37.
Verkhratsky, A., 2006b. Patching the glia reveals the functional
Yamanaka, K., Chun, S.J., Boillee, S., Fujimori-Tonou, N.,
organisation of the brain. Pflugers. Arch. 453, 411–420.
Yamashita, H., Gutmann, D.H., Takahashi, R., Misawa, H.,
Verkhratsky, A., 2009. Neuronismo y reticulismo: neuronal–glial
Cleveland, D.W., 2008. Astrocytes as determinants of disease
circuits unify the reticular and neuronal theories of brain
progression in inherited amyotrophic lateral sclerosis. Nat.
organization. Acta Physiol. (Oxf) 195, 111–122.
Neurosci. 11, 251–253.
Verkhratsky, A., Butt, A., 2007. Glial Neurobiology. A Textbook.
Yasojima, K., Schwab, C., McGeer, E.G., McGeer, P.L., 1999.
John Wiley & Sons, Chichester.
Distribution of cyclooxygenase-1 and cyclooxygenase-2
Verkhratsky, A., Kettenmann, H., 1996. Calcium signalling in glial
mRNAs and proteins in human brain and peripheral organs.
cells. Trends Neurosci. 19, 346–352.
Brain Res. 830, 226–236.
Verkhratsky, A., Kirchhoff, F., 2007a. Glutamate-mediated
Yasojima, K., Tourtellotte, W.W., McGeer, E.G., McGeer, P.L., 2001.
neuronal–glial transmission. J. Anat. 210, 651–660.
Marked increase in cyclooxygenase-2 in ALS spinal cord:
Verkhratsky, A., Kirchhoff, F., 2007b. NMDA receptors in glia.
implications for therapy. Neurology 57, 952–956.
Neuroscientist 13, 28–37.
Yoshihara, T., Ishigaki, S., Yamamoto, M., Liang, Y., Niwa, J.,
Takeuchi, H., Doyu, M., Sobue, G., 2002. Differential expression
Ferrante, R.J., Przedborski, S., Kristal, B.S., Friedlander, R.M.,
of inflammation- and apoptosis-related genes in spinal cords
2002. Minocycline inhibits cytochrome c release and delays
of a mutant SOD1 transgenic mouse model of familial
progression of amyotrophic lateral sclerosis in mice. Nature
amyotrophic lateral sclerosis. J. Neurochem. 80, 158–167.
417, 74–78.
Zerangue, N., Kavanaugh, M.P., 1996. Flux coupling in a neuronal
Zonta, M., Angulo, M.C., Gobbo, S., Rosengarten, B., Hossmann,
glutamate transporter. Nature 383, 634–637.
K.A., Pozzan, T., Carmignoto, G., 2003. Neuron-to-astrocyte
Zhu, S., Stavrovskaya, I.G., Drozda, M., Kim, B.Y.S., Ona, V., Li,
signaling is central to the dynamic control of brain
M.W., Sarang, S., Liu, A.S., Hartley, D.M., Du, C.W., Gullans, S.,
microcirculation. Nat. Neurosci. 6, 43–50.
Source: http://uem.avcr.cz/miranda2/export/sitesavcr/data.avcr.cz/lifesci/uem/research/publications/molecular-neurophysiology/files/heneka-2010.pdf
CSI – Birmingham Challenging Clinical Case Studies The Medical Model You Be the Detective! Important Studies, Work-up, and Protocols Alan Freint, MD Paul Pessis, AuD The Basic Premise Where to Start? Identifying the etiology of a health problem is essential
Pharmacology of Systemic Antibacterial Agents: Leena Palomo, DDS, MSD; Géza T. Terézhalmy, DDS, MA Continuing Education Units: 3 hours Online Course: Disclaimer: Participants must always be aware of the hazards of using limited knowledge in integrating new techniques or procedures into their practice. Only sound evidence-based dentistry should be used in patient therapy.







