Strokeindia.in
VASCULITIS
The vasculitides are a heterogeneous group of clinicopathological entities that share the common feature of vascular inflammation and injury. While a number of underlying causes can be identified in some disorders, the aetiology is unknown in many. The pathogenetic mechanisms involved are mainly immunological, immune complex mediated tissue injury being the most commonly incriminated factor. Vasculitis may be the sole manifestation of a disease or it may occur as part of another Primary disease. It may be confined to one organ or may affect multiple organs simultaneously.
Vasculitides is generally classified based on the size of the vessel involved:
I
. Large vessel vasculitis: Giant cell arteritis Takayasu's arteritis
II
. Mediumvessel vasculitis: Polyarteritis nodosa Kawasaki disease
III. Small vessel vasculitis: Microscopic polyangiitis Leukocytoclastic vasculitis Wegener's granulomatosis Churg-Strauss disease
Vasculitis Syndromes may also be classified as Primary and Secondary Vasculitis Syndromes.
ETIOLOGY & PATHOGENESIS
The cause remains undetermined in most vasculitic syndromes, while some forms of vasculitides may be ascribed to underlying factors like infections, drug reactions, malignancy or connective tissue disorders. Immunologic damage by immune-complex deposition or cell-mediated hypersensitivity is responsible in the majority of cases. A number of factors like genetic predisposition, environmental exposure and immue regulation to antigens are all important.
The possible immunopathologic mechanism in the causation of vasculitis are:
1
. Pathogenic immune complex formation and or deposition within the vessel wall. This leads to
complement activation and chemotactic attraction of neutrophils, subsequent phagocytosis
with liberation of neutrophil granular products leads to vascular damage. Eg. Henoch Schonlein purpura, Conective tisu diseases, Hepatitis B & C associated with Polyarteritis Nodosa and mixed ryglobulinemai.
2.
Cell-mediated hypersensitivity: Antigenic exposure may attract lymphocytes which liberate
cytokines causing tissue damage and further activation of macrophages and lymphocytes and
granuloma formation. Eg.Giant cell arteritis and Takayasu's Arteritis.
3. Production of Antineutrophilic Cytoplasmi Antibodies(ANCA) – Eg. Wegener's Granulomatosis, Churg Strauss Syndrome & Microscopic Polyangiitis.
ANCAs were originally described in Davies
et al in 1982 in segmental necrotising glomerulonephritis, and by van der Woude
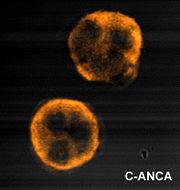
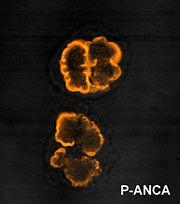
et al in 1985 in Wegener's. The Second International ANCA Workshop, held in The Netherlands in May 1989, fixed the nomenclature on perinuclear vs. cytoplasmic patterns, and the antigens MPO and PR3 were discovered in 1988 and 1989, respectively. ANCA are antibodies directed against antigens in the cytoplasmic granules of neutrophils and monocytes. Cytoplasmic ANCA (c-ANCA) refers to diffuse granular cytoplasmic staining seen under Indirect Immunofluorescent Microscope. The target antien is Proteinase 3(PR3), a 29-kDa neutral serine proteinase present in neutrophil azurophilic granules. More than 90% of patients with WG have c-ANCA. Perinuclear ANCA (p-ANCA) which is targeted against Myeloperoxidase(MPO), occur as a more localized perinuclear or nuclear staining pattern under IIF microscopy. Other antigens producing p-ANCA pattern include elastase, cathepsin G, lactoferrin, lysozyme and BPI. P-ANCA occur in MPA, CSS, GPS, Crescentic glomerulonephritis and also in WG.
Atypical ANCA(x-ANCA) with Snow-drift pattern in IIF, contains mixture of C- and P-ANCA pattern. These are nonspecific, directed against lactoferrin, lysozyme, beta-glucuronidase, cathepsin G, and so forth, found in Primary sclerosing cholangitis Primary biliary cirrhosis, Autoimmune hepatitis, SLE, RA, nonvasculitis conditions like Rheumatic and Nonrheumatic autoimmune diseases, inflammatory bowel disease, certain drugs, cystic fibrosis and bacterial endocarditis.
Microscopic
Wegener's
Churg-Strauss Syndrome
PR3-ANCA
MPO-ANCA
Negative
ANCA titres are generally measured using ELISA and indirect immunofluorescence.
Development of ANCA It is poorly understood. There is probably a genetic contribution, particularly in genes controlling the level of immune response. Two possible mechanisms of ANCA development are postulated:
Theory of molecular mimicry:Microbiaare molecules expressed by bacteria and
other microorganisms that have the power to stimulate a strong immune response by activation
of T-cells. These molecules generally have regions that resemble self-antigens – this is the theory
of molecular mimicry. Staphylococcal and streptococcal superantigens have been characterised
in autoimmune diseases – the classical example in post group A streptococcal rheumatic heart
disease, where there is similarity between M proteins of Streptococcus pyogenes to cardiac
myosin and laminin. It has also been shown that up to 70% of patients with Wegener's
granulomatosis are chronic nasal carriers of Staphylococcus aureus, with carriers having an eight
times increased risk of relapse.
Theory of defective apoptosis: Neutrophil apoptosis, or programmed cell death, is vital in
controlling the duration of the early inflammatory response, thus restricting damage to tissues by
the neutrophils. ANCA may be developed either via ineffective apoptosis or ineffective removal
of apoptotic cell fragments, leading to the exposure of the immune system to molecules normally
sequestered inside the cells.
Role in disease: It is unclear what the rôle of ANCA may be in these diseases – they may be markers of disease or may play some part in the pathogenic process. It has been shown that in Wegener's granulomatosis there is positive correlation between ANCA titre and disease activity and in vitro studies have shown that ANCA cause activation of primed neutrophils and react with endothelial cells expressing PR3. ANCA may act by causing release of lytic enzymes from the white blood cells, causing vasculitis.
Cytoplasmic (cANCA)
Perinuclear (pANCA)
Granular cytoplasmic staining
Homogenous perinuclear staining
Antigens: PR3 (90%), MPO (5%), & BPI (4%)
Antigens: MPO (70%), lactoferrin (10), elastase
(8%), cathepsin G (5%) & PR3 (2%)
cANCA disease association:
pANCA disease association:
Primary vasculitis
Primary Systemic Vasculitis
Wegener's granulomatosis
Microscopic polyangitis (MPO)
Microscopic polyangitis
Churg-Strauss syndrome (MPO)
Churg-Strauss syndrome
Polyarteritis nodosa (MPO)
Connective Tissue Diseases Felty's syndrome (lactoferrin)
Systemic lupus erythematosus (lactoferrin)
Rheumatiod arthritis (lMPO/lactoferrin) Sjögren's syndrome (MPO/lactoferrin)
Chronic inflammatory bowel disease Ulcerative colitis (cathepsin G/elastase) Crohn's disease (elastase)
Chronic liver disease Prim. Sclerosingcholangitis (cathepsinG)
PATHOLOGY:
For a definitive diagnosis of vasculitis, the presence of vascular damage, particularly in the form of fibrinoid degeneration, is essential. Perivascular cellular infiltration is a common histological finding. Vasculitis may involve blood vessels of varying calibers and this feature forms the basis of a useful pathological classification of vasculitis. An infiltrate, composed of a variety of cell types, like neutrophils, lymphocytes, and histiocytes may invade the vessel wall and the surrounding tissue. Extravasation of red cells is a prominent feature in many vasculitides. In Takayasu Arteritis and GCA Granulomatous inflammation with giant cell formation is a characteristic feature.
The signs and symptoms
The symptoms of vasculitis depend on which vessel and the particular organ involved by the inflammatory process. For example, GCA typically involves the medium– to large–sized blood vessels supplying the head and neck, but rarely involves the blood vessels of the kidneys. In contrast, Wegener's granulomatosis frequently involves the kidneys, very often the lungs, and almost always the upper respiratory tract, but rarely blood vessels to the brain. Buerger's disease involves the toes. Gangrene can result from a profound lack of blood flow.
General signs and symptoms of vasculitis experience include: Fever, fatigue, weight loss > 10%, muscle and joint pain and loss of appetite.Each type of vasculitis can also cause specific signs and symptoms.
Organ systems affected in Vasculitides: Skin:
A variety of rashes, the most classic of which is "palpable purpura" –purplish–red spots, usually found on the legs. These spots can usually be felt by the examiner's fingertips, hence the descriptor "palpable". They tend to occur in "crops".
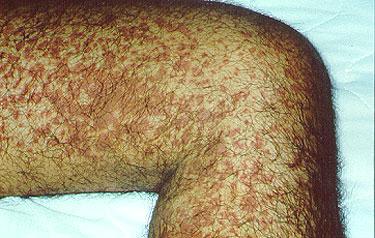
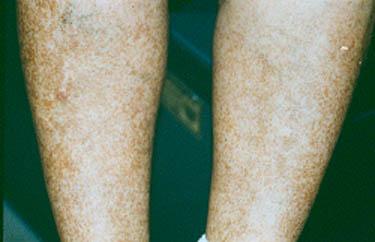
Repeated bouts of purpura may lead to hyperpigmented areas of the skin.
Symptoms range from full–blown arthritis to aches in the joints without obvious swelling (arthralgias).
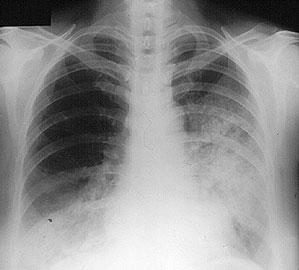
Lungs Cough, hemoptysis, shortness of breath, a pneumonia–like appearance to a patient's chest X–ray, lung "infiltrates", and the development of cavities in the lungs.
The X–ray shows fluffy infiltrates in both lungs, representing bleeding from damaged capillaries.
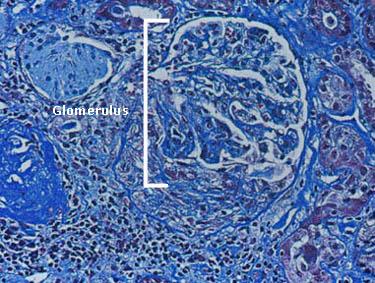
Microscopic hematuria, clumps of red blood cells (known as "casts"), and Proteinuria. Renal damae may lead to renal insufficiency and the requirement of dialysis. Depicted in the figure below is a single glomerulus (the filtering unit of the kidneys; each kidney has approximately 1 million glomeruli). This glomerulus is involved actively by an inflammatory process, particularly evident in the bottom of the figure.
Gastrointestinal Tract
Abdominal pain, bloody diarrhea, perforation of the intestines.
Blood Anemia (Normochromic, normocytic), a slightly elevated white blood cell count, elevated ESR and CRP.
Sinuses, Nose and Ears Chronic sinusitis that persist for longer than they should; hearing loss; inflammation of the nasal septum, sometimes resulting in a perforation or collapse of the bridge of the nose.
Eyes May affect either blood vessels to the eyes, causing the sudden loss of vision, or small blood vessels within the eyes, leading to retinal problems, thinning of the sclera, uveitis and conjunctivitis.
Headaches, strokes, changes in cognition, difficulty with coordination r locomotion.
Paraesthesias, pains in the arms and legs, numbness, and asymmetrical weakness (Mononeuritis Multiplex).
One of the least invasive ways of making the diagnosis. A minor procedure performed under local anesthesia. The wound is closed with 1–2 stitches that are removed 7–10 days later.
Skin biopsy showing leukocytoclastic vasculitis
WEGENER'S GRANULOMATOSIS(WG)
Wegener's granulomatosis (WG) is characterized by a triad of granulomatous necrotizing vasculitis of the upper and lower respiratory tracts, and glomerulonephritis.
Incidence & Prevalence
Uncommon with prevalence of 3/100,000 in the Western countries. Generally occur at the age of 40,although seen in children(15%).
Pathology & Pathogenesis
The histopathologic hallmarks of WG are necrotizing vasculitis of small arteries and veins with intra or extravascular granuloma formation in the respiratory tract and a rapidly progressive cresentic glomerulonephritis. Chronic nasal carriage of Staphylococcus aureus, is associated with higher relapse rate.There is an unbalancd TH1 type T cell cytokine profile.
Clinical & Laboratory manifestations
Upper airway disease(95%) with sinusitis, nasal obstruction, and bloody nasal discharge, nasal septal perforation with saddle nose deformity, subglottis stenosis,Cough, dyspnoea, and hemoptysis are common.
Eye signs include conjunctivitis, dacryocystitis, episcleritis, scleritis, granulomatous uveitis ad retroorbital masses withproptosis. Cutaneous features include palpable purpura, subcutaneous nodules, ulcers and vesiculo-bullous lesions. Pericarditis, coronary angiitis and cardiomyopathy can occur. Renal disease(77%) dominates and if left untreated, ends fatally. Hematuria, proteinuria, RBC casts with rapid onset of renal failure, unless appropriate treatment is started. CNS manifestations include polyneuritis cranialis, cerebral vasculitis or granuloma and mononeuritis multiplex.
WG may present with local or regional symptomatology without renal involvement (limited
Upper respiratory imaging study may show mucosal thickening, masses,
Pulmonary nodular or cavitary lesions, fixed infiltrates. Endobronchial or open lung biopsy shows necrotizing granulomatous vasculitis and extravascular granuloma.
Renal biopsy reveals Focal, segmental and crescentic pauci-immune glomerulonephritis.
The specificity of a positive antiproteinase-3 ANCA(cANCA) is very high and is a valuable
diagnostic tool in the diagnosis of WG. ANCA titres may reflect disease activity and a relapse may be preceeded with raising ANCA titres.
Differential Diagnosis include Midline Granuloma and lymphomatoid Granulomatosis.
Treatment: Once a rapidly fatal disease , the prognosis of WG has been greatly improved. Induction of
remission, maintenance of remission and prevention of relapses are the goals of treatment. Induction of
remission is achieved by Glucocorticoid ( Predisolone 1 mg/Kg per day initially, with gradual taper and
conversion to alternate day therapy after 6 monts) and cyclophosphamide therapy.( Oral 2 mg/Kg per
day). Intravenous pulses of cyclophosphamide are associated with increased relapse rates.
Glucocorticoid-related side effects include Diabetes Mellitus, cataracts, life threatening infections,
osteoporosis and cushingoid features. Alternate day regimens reduces toxicity. Cyclophosphamide-
related toxicities are frequent and severe. Cystitis in 30%, bladder cancer in 6%, Myelodysplasia in 2%
and infertility. After induction of remission with cyclophosphamide, to reduce the toxicity associated
with it, weekly low dose Methotrexate(7.5 – 15 mg./week) may be used for upto years after remission,
along with Folic acid, 1 mg daily, or folinic acid 5-10 mg once a week. Alternatively Azothioprine 2 mg/Kg
per day may be used. Mycophenolate mofetil, 1000 mg twice a day can be used in those patients who are unable to tolerate the above 2 drugs. Co-trimoxazole therapy may help in decreasing upper respiratory relapses.
CHURG-STRAUSS SYNDROME(CSS)
CSS also called Allergic Granulomatosis, it is a systemic granulomatous vasculitis which is characterized
by asthma, peripheral and tissue eosinophilia and extravascular granuloma formation.
Incidence & Prevalence
Rare, with an annual incidence of 1-3 per million. Mean age at onset is 48 years with mild female preponderance.
Pathology & Pathogenesis
It resembles PAN in many ways except that in CSS, the clinical features are dominated by pulmonary symptoms in the form of severe asthma and the histopathology shows granulomatous inflammation with prominent tissue eosinophilia.
Clinical & Laboratory manifestations
In addition to constitutional features like fever, malaise, and weight loss, there is severe, long-standing asthma and pulmonary infiltrates. Mononeuritis multiplex is common (72%). Cardiovascular lesions with myocarditis and congestive cardiac failure, and cutaneous lesions (nodules, purpura, hemorrhagic bulla) occur. Renal disease is less common and less severe than WG and MP.
The Characteristic laboratory finding is eosinophilia, which reaches > 1000cells/uL in >80% of patients. Anti-MPO ANCAis present in 48%.
Characteristic biopsy findings are a granulomatous vasculitis and extravascular granuloma with prominent eosinophilic infiltration. Severe peripheral blood eosinophilia, prominent pulmonary involvement with infiltrate and presence of pANCA is typical.
The prognosis is poor, with a 5 year survival of 25%. Systemic steroids with/without cyclophosphamide.
MICROSCOPIC POLYANGIITIS:
A necrotizing vasculitis with few/no immune compexes affecting small vessels
(capillaries, venules or arterioles), without granulomatous inflammation(1999,Chapel Hill Consensus Conference).
Incidence and Prevalence
Exact incidence unknown since it was previously included in Polyarteritis Nodosa. The mean age of onset is around 57 with male predominance.
Pathology and Pathogenesis
Microscopic polyangiitis(MPA) was considered to be a subset of PAN with involvement of small vessels (arterioles, venules, and capillaries) In contrast to PAN, in which renal involvement is due to arteritis, and pulmonary arteries are spared, MPA causes pauci immune glomerulonephritis like in Wegener's granulomatosis and alveolar capillaritis leading to pulmonary bleeding. It is commonly associated with the presence of pANCA ( perinuclear pattern of antineutrophil cytoplasmic antibody), which may play a role in pathogenesis.
Clinical and Laboratory manifestations
Similar to WG. Initial systemic symptoms like fever, followed byrapidly progressive glomerulonephritisin 79% of patients. Hemoptysis(12%), may be the first symptom. Upper respiratory and pulmonary symptoms are absent.
Elevated ESR,anemia, leukocytosis, thrombocytosis and Anti- MPO ANCA(p-ANCA)is present in 75%.
Presence of pauci-imune glomerulonephritis with above clinical picture.
The 5-year survival rate of treated patients is 74%. Treatment is with steroids and cyclophosphamide like in WG. Relapses occur in 34% of patients.
POLYARTERITIS NODOSA (PAN)
Also known as classic PAN, was described by Kussmaul and Maier in 1866. It is a
multisystem disease characterized histopathologically by segmental necrotizing vasculitis of medium and small-sized muscular arteries I hich renal and visceral arteies are involved in a characteristic pattern, involving the arterial bifurcations and resulting in aneurysm formation. PAN does not involve the pulmonary arteries.
Incidence and Prevalence
Uncommon disease, the exact prevalence of which is diffucult to establish.
PAN is commoner in men and the mean age of presentation is about 45 years.
Pathology and Pathogenesis
The histological findings in PAN is a segmental, necrotizing panarteritis of small and medium-sized arteries, characteristically involving the branching points. Initially polymorphpnuclear infiltrates and later mononuclear cell infiltrates with degeneration of vessel wall, intimal proliferation and fibrinoid necrosis results in thrombosis and infarction of the involved organs. Aneursymal dilatation upto 1 cm in size are characteristic of PAN. May be associated with Hepatitis B in 10-30% of patients, which suggests
the role of immunological phenomena in the pathogenesis of PAN. May also be associated with Hepatitis C infections, SLE, inflammatory bowel disease and hairy cell leukemia.
Clinical and Laboratory manifestations
The presentation is with constitutional symptoms like fever, weight loss, arthralgia, myalgia, and hypertension. The systems involved commonly are: the kidney
( hypertension, proteinuria, renal failure), GI tract ( pain, bleeding, nausea and vomiting), musculoskeletal system( arthralgia, arthritis and myalgia), peripheral nerves( peripheral neuropathy, mononeuritis multiplex), CNS (stroke), CVS (infarction) Skin ( subcutaneous nodules, purpura, infarcts, gangrene, livedo reticularis, Raynaud's) and genitourinary system ( testicular of ovarian pain). Pulmonary involvement is unusual.
Lab findings include: anaemia, raised ESR, leukocytosis, hypergammaglobulinemia, urinanalysis revealing proteinuria, red cells and casts. HBs antigen and ANCA(both p&c) may be positive.
Gangrene of toes
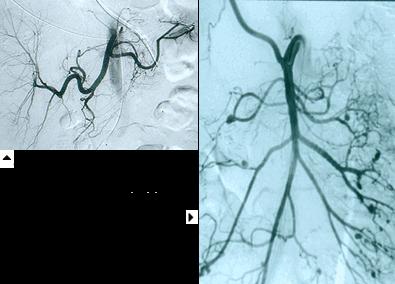
Based on biopsy. The preferable site of biopsy is an affected organ: skin, muscle or testis. If these sites are not involved, the diagnosis is based on angiography- which may reveal aneurysmal dilatations of renal, hepatic, or other visceral arteries.
The prognosis f untreated PAN is extremely poor, with 5-year survival rate of 10-20%. Death is due to GI bleeding or cardiovascular causes. Intractable hypertension adds to mortality and morbidity.Systemic corticosteroids like Prednisolone (1mg/kg/day)with gradual taper. Cyclophosphamide (2 mg/kg/day) may be given may be given with steroids. Once the disease is quiescent for about a year withdrawal of drugs may be tried. Relapses occur in about 10%. Antiviral therapy for Hepatitis B and antihypertensive drugs to control the blood pressure, prolongs survival.
TAKAYASU'S ARTERITIS (TA):
The first case of Takayasu's arteritis was described in 1908 by Dr. Mikito Takayasu. It is a serious condition that often presents with acute vascular occlusions or hypertension in young females. TA is a chronic inflammatory disease of the thoracic and abdominal aorta and its major branches and pulmonary arteries that most often affects young women in the second and third decades of life. Hence also called as Pulseless disease or aortic arch syndrome.
Incidence and Prevalence
TA is a rare form of primary systemic vasculitis that appears to be commoner in Asia than Europe or North America, where annual incidence rate is 1.2-2.6 cases per million. Females comprise 80-90% of patients with TA in most series, whereas in India, the Female preponderance is around 1.5: 1. Japanese patients with TA have a higher incidence of aortic arch involvement.
Pathology and Pathogenesis
TA has been reported in identical twins, leading to hypotheses of a hereditary basis for disease. In Japan and Korea, TA is associated with human leukocyte antigens HLA-A10, B5, Bw52, DR2, and DR4. These associations have not been confirmed in Western studies. TA is associated with HLA-B22 in the US. TA is characterized by granulomatous inflammation of the aorta and its major branches, leading to stenosis, thrombosis, and aneurysm formation. The lesions of TA are segmental with a patchy distribution. Mononuclear infiltration of the adventitia occurs early in the course of the disease, with cuffing of the vasa vasorum. Granulomatous changes may be observed in the tunica media with Langerhans cells and central necrosis of elastic fibers and smooth muscle cells. A panarteritis with infiltrates of lymphocytes, plasma cells, histiocytes, and giant cells is present. Later, fibrosis of the media and acellular thickening of the intima
compromise the vessel lumen. Stenoses are found in many of patients with TA disease. Often patients have poststenotic dilatations and other aneurysmal areas. Endothelial activation leads to a hypercoagulable state predisposing the patient to thrombosis. Congestive heart failure in individuals with TA may occur as a result of hypertension, aortic root dilation, or myocarditis. Transient ischemic attacks, cerebrovascular accidents, mesenteric ischemia, carotidynia, and claudication may occur. Symptoms of vascular compromise may be minimized by the development of collateral circulation with the slow onset of stenoses. Vessel wall dissection or aneurysm may occur in areas weakened by inflammation. One hypothesis for granulomatous vasculitis development is that antigens deposited in vascular walls activate CD4+ T cells, followed by release of cytokines chemotactic for monocytes. These monocytes are transformed into macrophages that mediate endothelial damage and granuloma formation in the vessel wall. Increased expression of intercellular adhesion molecule-1 (ICAM-1) and vascular cell adhesion molecule-1 (VCAM-1) is noted. Humoral immunity also is believed to be involved; antiaorta antibodies and antiendothelial cell antibodies have been found in patients with TA. Immunoglobulin G, immunoglobulin M, and properdin are found in lesions from pathologic specimens.
Histopathology of an arterial biopsy spicemen in TA
Clinical and Laboratory manifestations
The initial complaints may be nonspecific constitutional signs and symptoms like fever, malaise, weight loss and lethargy and hence the correct diagnosis may be delayed for months or years. Vascular insufficiency related to stenosis and thrombosis of affected vessels may cause renovascular hypertension, neurologic symptoms, or claudications. Cardiovascular system is the major organ involved with ascending aortitis leading to coronary ostitis causing myocardial ischemia or infarct, aortic
regurgitation, congestive heart failure, a widened mediastinum on chest roentgenogram which CT scan demonstrates as a dilated aortic arch. Despite the term pulseless disease, which is a synonym for TA, the predominant finding in individuals with TA is asymmetric pulse. Absent peripheral pulses occur late in the course of the disease. While 5-year survival rates exceed 90%, the disease has a high incidence of residual morbidity.
Disease monitoring in TA
Clinical analysis of TA disease activity, response to treatment and detection of relapse remains sub-optimal. Kerr and colleagues defined active TA as the new onset or worsening of 2 or more of the following features: fever or arthralgia; raised ESR (>20 mm/hr); features of vascular ischaemia or inflammation such as claudication, diminished or absent pulse, bruit, vascular pain, asymmetric blood pressure in upper or lower limbs or typical angiographic features; however this definition has not been widely validated and is limited in scope. The Birmingham Vasculitis Activity Score (BVAS), a reliable indicator of disease activity for primary systemic vasculitis, lacks sensitivity for the large vessel vasculitides and is rarely useful for clinical practice.
A new clinical index of disease severity and extent in TA (the DEI-TAK) which uses the BVAS index as its template. The index is based on the findings from 143 Indian patients with TA and contains 59 items in 11 organ-based scoring systems with emphasis on the cardiovascular system (19 items).
The ESR and CRP may be helpful in assessing disease activity and response to treatment.
Arteriography often demonstrates long, smooth, tapered narrowings or occlusions. Stenoses occur in 90-100% of patients with TA and aneurysm formation in only 27%. Based on Arterigraphy Numano et al have classified TA into 5 types:
Numano classification by Angiographic criteria
Carotid angiogram showing stenosis and Carotid angioplasty
and stenting of the internal carotid artery.
Arteriography. Typical features are: stenosis, post-stenotic dilatation, aneurysm, and increased collaterals. Three-dimensional MRA imaging of the aorta and its branches are providing exciting new data that may improve the understanding of the disease. Magnetic resonance imaging, magnetic resonance angiography, computed tomography : These examinations are useful for serial examinations and diagnosis in the early phase of TA. Ultrasound Duplex, CT scan and MRI may demonstrate mural thickening of the aorta and luminal narrowing. PET studies are a research tool.
High-resolution ultrasonography: showing normal flow in stented Carotid and Subclavian
Goals of medical therapy are to control active inflammation and to normalize clinical and laboratory parameters while preventing further vascular damage. Daily high-dose corticosteroid administration is the mainstay of initial therapy. Prednisolone at 1 mg/kg/d for 4-6 weeks, maintain high-dose treatment until all evidence of active disease has resolved, then taper prednisolone dosage gradually. Patients who relapse on corticosteroid taper may be controlled with weekly infusions of methylprednisolone (15 mg/kg, not to exceed 1 g/wk).
Immunosuppressive regimens including weekly methotrexate (7.5 -25 mg.) or daily or
monthly intravenous cyclophosphamide in individuals with glucocorticoid-resistant TA. Low-dose weekly methotrexate also has been used as a steroid-sparing agent for patients not tolerating corticosteroid taper. Cyclosporine may be an alternative therapy offering lower ovarian toxicity than cyclophosphamide. However, cyclosporine often is associated with decreased renal function and increased blood pressure, which may aggravate the damage to the heart and great vessels. Mycophenolate mofetil may be useful to treat individuals with glucocorticoid-resistant disease. TNF inhibition using etanercept or infliximab was successful in inducing clinical remission and permitting corticosteroid taper in patients who are steroid dependent. There are anecdotal reports of matrix metalloproteinase inhibition using minocycline.
Surgical Care: Following the acute phase, patients with fibrotic changes require surgical
treatment of symptomatic stenotic or occlusive disease. This can be achieved by percutaneous
angioplasty or stenting or, in severe cases, by resection and placement of a manmade graft.
Children with TA rarely require bypass surgery of carotid stenting. Percutaneous balloon
angioplasty of the aorta is reported to normalize systolic and diastolic blood pressures within 24 hours, with improvement of exercise tolerance and restoration of peripheral pulses. Both renovascular hypertension and congestive failure due to increased afterload are improved. Improvement has been sustained for as long as 3-5 years. Endovascular stenting is used in patients with severe stenoses, hypertension, or ischemia during the fibrotic phase of the disease. Multiple stents have been used in children to relieve long-segment renal artery stenosis and attendant renovascular hypertension.
GIANT CELL ARTERITIS (GCA)
Also referred to as cranial arteritis or temporal arteritis, it is typically a disease of elderly individuals. GCA is a granulomatous panarteritis with giant cell formation mainly involving branches of carotid artery, particularly the temporal artery, although the medium and large sized arteries at multiple locations may be involved.
Incidence and Prevalence
Occurs in individuals >50 years, with annual incidence rates of 6.9 to 32.8 per 100,000 population. Commoner in women and rare in blacks. Familial aggregation and association with HLA-DR4 and HLA-DRB1*O4 have been reported. Polymyalgia rheumatica(PMR) with an annual incidence of 58.7 per 100,000 population, is commonly associated with GCA.
Pathology and Pathogenesis
Histopathologically, there is panarteritis with inflammatory mononuclear cell infiltrates within the vessel wall with frequent giant cell formation, proliferation of intima and fragmentation of internal elastic lamina leading to vessel stenosis and tissue ischemia. An antigen in arterial wall is recognized by T cells, with production of IL-2, IL-6 and IFN-gamma.
Clinical and Laboratory manifestations
Typical features are fever, severe unilateral headache, anemia and raised ESR. The affected temporal artery becomes thick, tortuous, and tender. Scalp pain and claudication of jaw and tongue may occur. Retinal arterial involvement ( anterior and posterior ischemic optic neuropathy) may lead to blindness,
if early diagnosis and prompt steroid therapy is not instituted. Claudication of extremities, strokes, myocardial infarctions and aortic aneurysms, have all been reported.
PMR is characterized by stiffness, aching and pain in the muscles of the neck, shoulders, back, hips and thighs.
Lab findings include elevated ESR, normocytic anemia and increased serum alkaline phosphatase, IgG and complement .
Based on characteristic constellation of clinical findings with raised ESR in elderly patients. Ultrasound Duplex scan of the Superficial temporal arteries reveal typical ‘Halo sign'. A Temporal artery biopsy should be done as quickly as possible. A biopsy segment of 3-5 cm with serial sectioning is essential, since segmental involvement is common in GCA.
GCA is highly responsive to systemic steroid therapy. Treatment should be is started early with Prednisolone 40-60 mg per day for 1 month, with gradual tapering. Patients require treatment for > 2 years, with ESR monitoring. Methotrexate has been used as steroid-sparing agent along with prednisolone.
HENOCH-SCHONLEIN PURPURA (HSP)
Also referred to as Anaphylactoid purpura, is a distinct small vessel vasculitic syndrome that is characterized by palpable purpura, commonly distributed over the buttocks and lower limbs, arthralgias, abdominal pain and occasionally glomerulonephritis.
Incidence and Prevalence
Usually seen in children between 4-7 years. Commoner in males with a peak incidence in Spring.
Pathology and Pathogenesis
Vascular deposition of IgA immune complexes is responsible for this multisystem disorder. A number of underlying factors has been associated including upper respiratory tract infections, insect bites, drugs( penicillin, erythromycin, chrorpromazine), infections( varicella, hepatitis B, Streptococcus), vaccinations ( measles, typhoid) and various malignancies.
Clinical and Laboratory manifestations
Children aged 2-14 years are typically affected, but may occur at any age. The hallmark of the disease is palpable purpura, usually occurring on the lower legs, ankle, buttock and thigh. Abdominal pain (70%) and bleeding, bowel perforation and infarction and intussusception may also occur. Arthralgia and arthritis mainly involve the ankle and knees. Renal involvement occurs in 10-50% with hematuria, proteinuria and chronic renal failure is rare in children. Adults have more frequent renal and myocardial involvement. Less common manifestations include testicular pain, seizure, mononeuritis multiplex and pulmonary hemorrhage.
Lab studies show leucocytosis, and elevated IgA in 50% of patients.
Based on clinical findings and histology of skin biopsy specimen and direct immunofluorescence test showing perivascular deposition of IgA and C3, confirm the diagnosis. Renal biopsy is rarely required to provide prognostic information in some patients.
Usually a self-limiting disease, 1-5% of children progress to end-stage renal disease. Symptomatic therapy is only needed. Systemic steroids are necessary for nephrotic range proteinuria and severe abdominal symptoms. Prednisolone, is started in dose of 1 mg per kg per day and tapered according to clinical response. Periodic urinalysis is advised to monitor renal involvement and there are anecdotal reports of benefits from plasmapharesis combined with cytotoxic drugs. 10-40% of patients have recurrences.
IDIOPATHIC CUTANEOUS VASCULITIS
Also known as allergic angiitis, leukocytoclastic angiitis, hypersensitivity vasculitis and cutaneous necrotizing venulitis. The clinical hallmark of cutaneous small vessel vasculitis is palpable purpura. The disease is most often induced by immune complex deposition and subsequent complement activation. It may be idiopathic or may be caused by a number of underlying primary systemic vasculitis or drugs.
Incidence and Prevalence
Represents the most commonly encountered vasculitis in clinical practice.
Pathology and Pathogenesis
It is characterized by segmental vasculitis of small vessels (particularly post-capillary venules) with endothelial swelling, fibrinoid necrosis of vessel wall and a prominent neutrophilic infiltrate with fragmentation of their nuclei (leukocytoclasia). RBCs extravasate from the involved vessels, leading to palpable purpura.
Clinical and Laboratory manifestations
Constitutional symptoms: Fever, anorexia, myalgia and Polyarthralgia.
Cutaneous: Palpable purpura, recurrent or persistent urticaria, papulonodular lesions, Vesicles and bullae, hemorrhagic pustules, infarcts and ulcers. Occur in lower extremities or in the sacral region of bedridden patients.
Extra-cutaneous: Joint pain specially in the lower limbs, renal dysfunction is rare. Abdominal symptoms: nausea, vomiting, pain abdomen and GI bleeds. Ankle edema and peripheral neuropathy may be seen.
Lab tests should be aimed towards ruling out underlying connective tissue disease or systemic vasculitis.
Cutaneous Vasculitis in left leg and dorsum of foot
Made by Skin biopsy which reveals characteristic features of leukocytoclasia. Immunofluorescence studies may show deposition of immunoglobulins and complement fractions around the blood vessels.
Removal of the antigen, antibiotics for infections and treatment of the underlying systemic disease are essential. For milder form of the disease with skin manifestations only, a period of rest, analgesics and antihistaminics is enough. For more severe cutaneous forms Hydroxychloroquine, dapsone or colchicine may be tried. Prominent systemic and cutaneous changes not responding to above may require corticosteroids and/or immunosuppressants like cyclophosphamide, azathioprine or methotrexate.
ESSENTIAL MIXED CRYOGLOBULINEMIA
Also known as cryoglobulinemia, cryoproteinemia and mixed cryoglobulinemia. Cryoglobulins are single or mixed immunoglobulins that undergo reversible precipitation at low temperatures. Cryoglobulinemia is characterized by the presence of cryoglobulins in the serum which may result in a clinical syndrome of systemic inflammation caused by cryoglobulin-containing immune complexes.
Incidence and Prevalence
Rare disease. The female-to-male ratio is 3:1, with the mean age at 42-52 years. The prevalence of mixed cryoglobulinemia is related to the endemic presence of HCV infection. Brouet et al (1974) reported the following frequencies: type I, 25%; type II, 25%; and type III, 50%.
Pathology and Pathogenesis
Cryoglobulinemia without an associated disease is known as idiopathic or essential, cryoglobulinemia and that associated with an underlying illness (lymphoproliferative disorder, autoimmune disease, infectious disease) as secondary cryoglobulinemia.
Cryoglobulinemia may be classified based on cryoglobulin composition with the Brouet classification: Type I cryoglobulinemia, or simple cryoglobulinemia - the result of a monoclonal immunoglobulin, usually immunoglobulin M (IgM) or, less frequently, immunoglobulin G (IgG), immunoglobulin A (IgA), or light chains. Types II and III cryoglobulinemia (mixed cryoglobulinemia) contain rheumatoid factors (RFs), which are usually IgM and, rarely, IgG or IgA. These RFs form complexes with the fragment, crystallizable (Fc) portion of polyclonal IgG. The actual RF may be monoclonal (in type II cryoglobulinemia) or polyclonal (in type III cryoglobulinemia) immunoglobulin. Types II and III cryoglobulinemia represent 80% of all cryoglobulins.
The mechanisms of cryoprecipitation : The solubility of cryoglobulins has been found to be partially related to the structure of component immunoglobulin heavy and light chains. Alteration in protein conformation with temperature changes leads to decreased solubility and subsequent vasculitic damage. Immune-complexes lead to glomerulonephritis, intravascular cryoglobulin deposits, reduced levels of complement, and complement fragments (C3a, C5a) that act as chemotactic mediators of inflammation. Cryoprecipitation leads to thrombosis of small arteries and capillaries in the extremities (gangrene) and glomeruli acute renal failure. Circulating large molecular weight cryoprotein complexes also produces Histologicallyis almost always the lesion in mixed cryoglobulinemia.
Type I cryoglobulins are usually monoclonal IgM, is typically related to an underlying lymphoproliferative disease.Types II and III, also known as the mixed cryoglobulinemias, are associated with chronic inflammatory states such as SLE and viral infections (particularly HCV). In these disorders, the IgG fraction is always polyclonal with either monoclonal (type II) or polyclonal (type III) IgM (rarely IgA or IgG) with RF activity (ability to bind IgG).
Clinical and Laboratory manifestations Meltzer triad, ie, purpura, arthralgia, and weakness, was first described in 1966 by Meltzer and Franklin in cases of essential mixed cryoglobulinemia. This triad is generally seen with types II and III cryoglobulinemia and is seen in up to 25-30% of patients.
Clinical manifestations include, General like fever, fatigue, weight loss.Cutaneous: lesions have a predilection for dependent areas (particularly the lower extremities) and include erythematous macules and purpuric papules (90-95%), as well as ulcerations (10-25%). Lesions in nondependent areas are more common in type I cryoglobulinemia (head and mucosa), as are livedo reticularis, Raynaud phenomenon, and ulcerations. Nailfold capillary abnormalities are common and include dilatation, altered orientation, capillary shortening, and neoangiogenesis.Musculoskeletal (70%): arthralgias and myalgias are rare in type I cryoglobulinemia and are common in types II and III disease. arthralgias in the proximal interphalangeal [PIP] joints, Metacarpophalangeal [MCP] joints, knees, and ankles)Frank arthritis and myositis are rare. Arthralgias commonly affect the proximal interphalangeal and metacarpophalangeal joints of the hands, knees, and ankles. Renal(5-60%): Clinicaland hematuria are more common than or acute renal failure. Renal involvement is one of the most serious complications of cryoglobulinemia and typically manifests early in the course of the disease (within 3-5 y of diagnosis). Failure to treat may result in renal failure.Pulmonary: A reduction in forced expiratory flow rates and the presence of interstitial infiltrates revealed by chest radiographs are common in mixed cryoglobulinemia. Approximately 40-50% of patients are symptomatic with dyspnea, cough, or pleuritic pain. Severe pulmonary disease is rare.Neuropathy(70-80%): Neuropathy is common in types II and III disease (as determined with electromyographic and nerve conduction studies).Sensory fibers are more commonly affected than motor fibers, with pure motor neuropathy in approximately 5% of patients. Abdominal pain(2-22%): Vasculitis of the small mesenteric vessels leads to acute abdomen. Sicca symptoms(4-20%), Acrocyanosis(9%),Arterial thrombosis(1%) of patients. Nervous system manifestations Sensorimotor neuropathy Visual disturbances CNS involvement (rare, although pseudotumor cerebri and cerebral vascular events have been described)
Evaluation for serum cryoglobulins at one and seven days at 4 degree C., Spectrophotometric analysis and specific immunologic assays may be used to identify cryoglobulin components (immunoglobulins, light chains, clonality). Urinalysis may represent evidence of renal disease, Complete blood cell count: Leukocytosis, anemia, elevated serum creatinine levels and electrolyte abnormalities. Liver function studies may reveal evidence of underlying hepatitis; obtain hepatitis serology. RF: RF is positive in types II and III. ANA is indicated upon clinical suspicion of underlying connective-tissue disease (SLE, Sjögren syndrome). Erythrocyte sedimentation rate (ESR): Elevations may be secondary to rouleaux formation. Complement evaluation (CH50, C3, C4): Patients may display hypocomplementemia (especially low C4 levels). Serum protein electrophoresis (SPEP), urine protein electrophoresis (UPEP), and quantitative immunoglobulin upon suspicion for underlying gammopathy. Serum viscosity if symptoms warrant. Imaging Studies: A chest radiograph may reveal interstitial involvement or pleural effusions, CT imaging may be considered upon high suspicion of underlying malignancy, Transesophageal echocardiography should be obtained if bacterial endocarditis is suspected, Angiography may be considered to evaluate for evidence of vasculitis.
Tissue biopsy may be required for diagnosis when patients with vasculitis, renal disease, or both are evaluated, Electromyography and nerve conduction studies may be used to confirm neuropathy and
further diagnostic procedures (eg, bone marrow biopsy, liver biopsy) usually depend on coexistent disease, especially HCV infection.
Histologic Findings: Skin: Purpura are histologically characterized by dermal vasculitis that extends variably to the subcutaneous interstitial space. HCV-associated proteins have been found in vasculitic skin biopsy samples. Autopsy studies have revealed unsuspected vasculitis of multiple organs (heart, lung, gastrointestinal tract, central nervous system, liver, muscle, adrenals). Histologic evaluation of affected lung, kidney, and muscle reveals eosinophilic material in the lumen of small vessels with frequent extension into the vessel intima and inflammation of the vessel wall.
The overall prognosis is worse in persons with concomitant renal disease, lymphoproliferative disease, or plasma cell disorders. Mean survival is approximately 50% at 10 years after diagnosis. Survival rates among patients with renal involvement is 60% at the end of 5 years.
The goal of therapy is to treat underlying conditions, as well as to limit the precipitant cryoglobulin and the resultant inflammatory effects. Asymptomatic cryoglobulinemia does not require treatment. Secondary cryoglobulinemia is managed with treatment of the underlying malignancy or associated disease. NSAIDs may be used for arthralgia and fatigue. Immunosuppressive medications (eg, corticosteroid therapy and/or cyclophosphamide or azathioprine) are indicated for vasculitis, renal disease, progressive neuropathy, or disabling skin manifestations. Plasmapheresis is indicated for severe or life-threatening Pegylated interferon alfa (IFN-alfa) combined with ribavirin has demonstrated efficacy in patients with cryoglobulinemia associated with hepatitis C, and efficacy in patients with chronic myelogenous leukemias and low-grade lymphomas has been reported. Anti-CD20 chimeric monoclonal antibody rituximab is effective in controlling disease manifestations such as vasculitis, peripheral neuropathy, arthralgias, low-grade B-cell lymphomas, renal disease, and fever.Rituximab therapy has been used predominately in HCV-related mixed cryoglobulinemia refractory to or unsuitable for corticosteroids and antiviral (IFN-alfa) therapy. Rituximab therapy is reportedly well tolerated in this patient population; however, treatment results in increased titers of HCV-RNA of undetermined significance.
CENTRAL NERVOUS SYSTEM VASCULITIS
Primary angiitis of the central nervous system is a rare disease characterized by vasculitis of small and medium arteries in the brain and spinal cord. Presentations are highly variable, but the triad of headache, organic brain syndrome, and multifocal neurologic deficits is most suggestive. Most affected patients do not have systemic symptoms, signs, or abnormal routine blood tests, including ESR.
Types of CNS Vasculitis
It may be classified as primary (primary angiitis of the central nervous system (PACNS) or isolated
vasculitis of the CNS when there is no other disease or condition causing vasculitis, or Secondary
vasculitis of the CNS system which is more common and can be a manifestation of autoimmune diseases
such as systemic lupus erythematosus (SLE), systemic necrotizing vasculitides like Wegener's
granulomatosis, Polyarteritis nodosa, or may be induced by drugs like amphetamines or cocaine.
Types of PACNS: Primary granulomatous angiitis (GACNS) of the CNS and benign angiopathy of the CNS (BACNS) also referred to as reversible vasoconstrictive (vessel spasm) disease.
GACNS: The distribution is nearly equally between the sexes, with perhaps a slight male predominance (4:3). The mean age of people affected by the disease is approximately 42 years, but the range is wide: the disease has been detected in children as young as 3, and also frequently occurs in elderly people.
BACNS: Young women, often those with previous histories of headaches are affected. They often have histories of heavy nicotine or caffeine use, over the counter cold remedy use (e.g., ephedrine), and oral contraceptive or estrogen replacement therapy, but the precise relationship (if any) of these exposures to the development of BACNS remains unclear.
The signs and symptoms of CNS Vasculitis
Onset may be sudden, subacute or a waxing and waning illness where there are multiple neurologic
signs and symptoms which evolve over many weeks or months. There may be transient ischemic attacks
or brief periods of visual loss, inability to use an arm or a leg or speech impairment. Severe headache
that is unresponsive to conventional therapy is also a very common symptom. Occasionally spinal cord
may be involved with weakness of the arms and legs. Less specific symptoms are memory loss,
dementia, organic brain syndrome or incontinence of bladder or bowels.
The symptoms of CNS vasculitis may be extremely difficult to separate from common neurologic disease like strokes, infections or even multiple sclerosis.
Diagnosis Perhaps no other form of vasculitis is as difficult to diagnose as CNS vasculitis. Tests that may provide clues, but not a certain diagnosis include cerebral spinal fluid analysis which reveals a high mononuclear cell count in GACNS, CT /MRI with MRA scans and four vessel digital subtraction Cerebral arteriography. Biopsy of the brain and leptomeninges, is the most direct means of making the diagnosis of CNS vasculitis.
Because angiography is less invasive than a brain biopsy, this test is often performed before biopsy. The classic angiographic findings in CNSV include "beading" (alternating dilatations and narrowings of blood vessels), aneurysms, and other irregularities within blood vessels. It must be recognized, however, that many conditions not caused by vasculitis (e.g., spasm of the blood vessels) can cause an angiographic appearance that is impossible to distinguish from true vasculitis.
MR imaging is very sensitive for CNS vasculitis and typically shows supratentorial infarctions in the cortical and subcortical regions; however, the MR appearance is not specific for CNS vasculitis and the correlation between MR imaging and angiography on specific lesions is moderate. The discrepancies between MR imaging and angiography are due to: 1. Different times at which the studies were performed 2. A patient with vascular narrowing but no tissue infarction might have an angiogram with positive results but an MR image with negative results. 3. Small-vessel abnormalities not apparent on an angiogram might lead to infarction detectable by MR imaging 4. Subtle MR abnormalities may escape detection unless special sequences or orientations are obtained.
Cerebral angiography often provides additional information about the extent of disease beyond that detected by MR imaging. The response to treatment is more likely to be evident on angiograms than on MR images. Administration of contrast material increases the detection of small infarctions and is also useful in estimating the age of lesions. Other imaging techniques for CNS vasculitis include perfusion- and diffusion-weighted imaging, MR spectroscopy, and positron emission tomography.
MRI of Brain showing infarction in Pons and Cerebellum
Cerebral Angiograms showing lesions in: 1. Right M1 (A, arrows) 2. left A2 and M2 (B, long and short arrows, respectively)3. In both PCAs (C, short arrows)
Because the diagnosis cannot be proven with 100% certainty by angiography, consideration is often given to performing a brain biopsy. If no obvious site for biopsy is identified by non–invasive studies or by angiography, the brain biopsy is usually performed on the non–dominant side of the patient's brain. Biopsy of the meninges, is usually performed at the same time (this increases the chance that the procedure will yield a piece of tissue containing pathology). Although brain biopsy remains the "gold standard" in the diagnosis of CNSV, 25% of the time a brain biopsy will be negative even in the setting of true vasculitis.
Many conditions can mimic vasculitis. These mimics have completely different treatments e.g. antibiotics for infections or surgery or radiation therapy or chemotherapy for brain tumors. Thus it is essential to rule out such diagnoses before embarking upon therapy for CNS vasculitis.
PACNS: Till recently, CNS vasculitis was a fatal condition, with death following in a mean of 45 days after diagnosis. The availability of powerful immunosuppressive therapy, however, has significantly improved the prognosis. Some respond well to treatment with high doses of steroids alone. Others require the addition of cyclophosphamide. A reasonable approach is to attempt to control the disease with high doses of steroids first (e.g., for one month), adding cyclophosphamide only if steroids fail or if patients begin to develop unacceptable side effects of steroid treatment. For PACNS, treatment must often be continued for a year or more.
BACNS: these patients require less intensive treatment regimens and may be treated with calcium–channel blockers like Verapamil or Nimodipine for a few weeks, along with a comparatively short course of steroids (prednisone). No firm guidelines exist regarding the length of therapy.
Possible side effects of high doses of glucocorticoids include diabetes mellitus, hypertension, weight gain, osteoporosis and increased risk of infections.
Cyclophosphamide induced toxicities include leucopenia, increased susceptibility to infections, cystitis, bladder cancer and a higher incidence of lymphomas later in life. Regular blood counts and urinalysis are essential. It is generally advised that patients receive antibiotic prophylaxis to prevent infections like pneumocystic carinii pneumonia, especially during the first few months of therapy.
Kawasaki disease (KD)
Also known as muco cutaneous lumph node syndrome, is an acute, self-limited vasculitis of unknown etiology that occurs predominantly in infants and young children. First described in Japan in l967 by Tomisaku Kawasaki, the disease is now known to occur in both endemic and community-wide epidemic forms. Kawasaki disease is characterized by fever, bilateral nonexudative conjunctivitis, erythema of the lips and oral mucosa, changes in the extremities, rash, and cervical lymphadenopathy. Coronary artery aneurysms or ectasia develop in 15% to 25% of untreated children and may lead to myocardial infarction (MI), sudden death, or ischemic heart disease.
Incidence and Prevalence
More prevalent in Japan, with an annual incidence of 112 cases per 100 000 children <5 years old and recurrence rate of 3%. Occurrence in siblings and twins, and the occurrence of KD in children of parents who themselves had the illness in childhood supports the contribution of genetic factors.
Pathology and Pathogenesis The etiology of Kawasaki disease remains unknown. The genetic basis of susceptibility is currently unknown. Kawasaki disease may be related to a bacterial superantigenic toxin, as suggested by selective expansion of Vß2 and Vß8 T-cell receptor families. The immune response in Kawasaki disease is oligoclonal rather than polyclonal, and immunoglobulin A (IgA) plasma cells play a central role. The key steps leading to coronary arteritis are: endothelial cell activation by CD68+ monocyte/
macrophages, CD8+ (cytotoxic) lymphocytes, and oligoclonal IgA plasma cells. Enzymes including matrix metalloproteinases that are capable of damaging arterial wall integrity may be important in the development of aneurysmal dilatation. Vascular endothelial growth factor (VEGF), monocyte chemotactic and activating factor (MCAF or MCP-1), tumor necrosis factor- (TNF- ), and various interleukins play important roles in the vasculitic process. Kawasaki disease is a generalized systemic vasculitis with aneurysms in mesenteric, femoral, iliac, renal, axillary and brachial arteries. In the early stages, the media demonstrate edematous dissociation of the smooth muscle cells, endothelial cell swelling and subendothelial edema,, influx of neutrophils is found in the early stages (7 to 9 days after
onset), with a rapid transition to large mononuclear cells in concert with lymphocytes (predominantly CD8+ T cells) and IgA plasma cells. Destruction of the internal elastic lamina and eventually fibroblastic proliferation occur at this stage. Active inflammation is replaced over several weeks to months by progressive fibrosis, with scar formation. Arterial remodeling or revascularization may occur in Kawasaki disease with coronary arteritis. Progressive stenosis results from intimal proliferation and neoangiogenesis. Lymphadenopathy, an early finding,usually disappears by autopsy. Pathological findings in lymph nodes include thrombotic arteriolitis and severe lymphadenitis with necrosis.
Clinical Findings and Laboratory manifestations
Classification Criteria for KD:
I. Fever persisting for at least 5 days.
II. Presence of at least 4 principal features: 1. Changes in extremities: Acute: Erythema of palms, soles; edema of hands, feet; Subacute: Periungual peeling of fingers, toes in weeks 2 and 3, Polymorphous exanthema. 2. Bilateral bulbar conjunctival injection without exudates. 3. Changes in lips and oral cavity: Erythema, lips cracking, strawberry tongue, diffuse injection of oral and pharyngeal mucosae. 4. Cervical lymphadenopathy (>1.5-cm diameter), usually unilateral.
Exclusion of other diseases with similar findings.
Patients with fever at least for 5 d and <4 principal criteria can be diagnosed with Kawasaki disease when coronary artery abnormalities detected by 2-D echocardiography or angiography. In the presence of 4 principal criteria, diagnosis can be made on day 4 of illness.
Cardiovascular findings Congestive heart failure, myocarditis, pericarditis, valvular regurgitation Coronary artery abnormalities like aneurysms of medium-size noncoronary arteries, Raynaud's phenomenon, Peripheral gangrene, Musculoskeletal system features like arthritis, arthralgia, Gastrointestinal tract manifestations like Diarrhea, vomiting, abdominal pain, Hepatic dysfunction, Hydrops of gallbladder, Central nervous system involvement includes extreme irritability, aseptic meningitis, Sensorineural hearing loss, Genitourinary system features include Urethritis/meatitis. Anterior uveitis and desquamating rash in groin may be present.
Laboratory findings: Leukocytosis with neutrophilia and immature forms, elevated ESR & C-reactive protein, Anemia, Abnormal plasma lipids, Hypoalbuminemia Hyponatremia, Thrombocytosis after week 1, Sterile pyuria, elevated serum transaminases & gamma glutamyl transpeptidase, pleocytosis of cerebrospinal fluid and Leukocytosis in synovial fluid.
Incomplete (Atypical) Kawasaki Disease
Patients who do not fulfill the criteria are diagnosed as having "incomplete" or "atypical"
Kawasaki disease, based on echocardiographic findings of coronary artery abnormalities.
Coronary Angiogram reveals coronary aneurysms in the proximal LAD and proximal RCA, followed by the LMCA, then LCX, and finally the distal RCA and the junction between the RCA and posterior descending coronary artery. When a coronary artery is larger than normal (dilated) without a segmental aneurysm, the vessel is considered ectatic. Intravascular ultrasound (IVUS), transesophageal echocardiography, and other modalities including magnetic resonance angiography (MRA) and ultrafast computed tomography (CT) may be of value in the assessment of selected patients.
Coronary angiogram demonstrating giant aneurysm of the LAD with obstruction
and giant aneurysm of the RCA with area of severe narrowing in 6-year-old boy.
Kawasaki disease is the leading cause of acquired heart disease in children in the United States. Coronary artery aneurysms or ectasia develop in 15% to 25% of untreated children; treatment with IVIG in the acute phase of the disease reduces this risk to <5%. During the acute phase of illness, aspirin is administered at 80 to 100 mg/kg per day in 4 doses for 14 days, followed by loer doses with IVIG (2 gm/Kg as a single infusion over 10-12 hours). Pentoxifylline is a methyl xanthine compound that specifically inhibits TNF- messenger RNA transcription. Because TNF- appears to be important in the inflammatory cascade in Kawasaki disease, pentoxifylline has been assessed as a therapeutic adjunct to standard therapy. Approximately 10% of patients who fail to defervesce with initial IVIG therapy, benefit from retreatment with IVIG along with Corticosteroids( intravenous pulse methylprednisolone, 30 mg/kg for 2 to 3 hours, administered once daily for 1 to 3 days. Plasma exchange, because of its risks, is not in generally recommended. Ulinastatin is a human trypsin inhibitor purified from human urine that has been used in Japan as an adjunctive therapy for acute Kawasaki disease. Abciximab, a platelet glycoprotein IIb/IIIa receptor inhibitor, has been used to treat patients in the acute or subacute phase of Kawasaki disease who have large coronary aneurysms. A humanized monoclonal antibody against TNF- , infliximab, might be considered in patients who are resistant to IVIG and steroids. Cytotoxic agents such as cyclophosphamide, in conjunction with oral steroids, have been suggested as useful for the treatment of exceptional patients with particularly refractory acute Kawasaki disease.
BEHCET'S SYNDROME
It is characterised by recurrent episodes of oral and genital ulcers, iritis, and cutaneous lesions. The underlying pathology is leukocytoclastic venulitis.
COGAN'S SYNDROME
It is a systemic vasculitis characterised by interstitial keratitis with vestibuloauditory symptoms. There is aortitis with involvement of aortic valve. Glucocorticoids are the mainstay of treatment, immediate initiation following the onset of hearing loss results in a favourable outcome.
POLYANGIITIS OVERLAP SYNDROMES
Some patients with systemic vasculitis manifest characteristics that do not fit into specific disease but have overlapping features of different vasculitides. The diagnostic as well as therapeutic considerations, prognosis for these patients depends upon site and severity of active vasculitis. Patients with vasculitis capable of causing irreversible organ system damage should be treated as described under Wegner's granulomatosis.
SECONDARY VASCULITIS
Drug-induced Vasculitis
These present as palpable purpura either generalised or limited to lower extremities. Urticaria, ulcers and hemorrhagic blisters may also occur. Signs and symptoms include fever, malaise and polyarthralgia. Skin is predominantly involved. Drugs that have been implicated are allopurinol, thiazides, gold, sulphonamides, phenytoin and penicillin. An increasing number of drugs are reported to cause vasculitis associated with antimyeloperoxidase ANCA, most evidently hydralazine and propylthiouracil. Manifestations range from cutaneous lesions to glomerulonephritis and pulmonary hemorrhage. The offending drug should be stopped immediately and treated with glucocorticoids and cyclophosphamide, if life threatening complications occur.
Serum Sickness and Serum Sickness-Like Reactions
These are characterised by fever, urticaria, polyarthralgia and lymphadenopathy 7 to 10 days after primary exposure and 2 to 4 days after secondary exposure to heterologous protein or a non protein drug like penicillin or sulfa. Patients may have cutaneous venulitis that may progress rarely to a systemic vasculitis.
Vasculits Associated with Other Underlying Primary Diseases
Certain infections may trigger an inflammatory vasculitis. Rickettsias can invade and proliferate in the endothelial cells of small blood vessels causing vasculitis. Fungal infections include histoplasmosis. Leukocytoclastic vasculitis involving the skin may be seen in subacute bacterial endocarditis, Epstein-Barr virus infection and HIV infection. Vasculitis associated with malignancies may be lymphoid or reticulo-endothelial neoplasms where leukocytoclastic venulitis is the most common finding. Hairy cell leukemia is associated with PAN.
Vasculitis as a secondary manifestation in connective tissue diseases is seen in SLE, Rheumatoid arthritis, inflammatory myositis, relapsing polychondritis and Sjogren's syndrome.
Secondary vasculitis is also associated with ulcerative colitis, congenital deficiencies of complement, retroperitoneal fibrosis, primary biliary cirrhosis, alpha1 antitrypsin deficiency and intestinal bypass surgery.
Source: http://strokeindia.in/pdf/vasculitis.pdf
Diagnosis, Treatment and Monitoring of Hyperadrenocorticism CAUTION: Federal (USA) law restricts this drug to use by or on the order of a licensed veterinarian. As with all drugs, side effects may occur. In field studies and post- approval experience, the most common side effects reported were: anorexia, lethargy/depression, vomiting, diarrhea, elevated liver enzymes, elevated potassium with or
Section II – Abstracts Process Monitoring, Control & Sensors PPS-24 – The Polymer Processing Society 24th Annual Meeting – June 15-19, 2008 Salerno – Italy Section II – Abstracts Terahertz Spectroscopy – A New Tool for Monitoring Compounding Processes Hochrein Thomas 1, Krumbholz Norman 2, Kretschmer Karsten 1, Bastian Martin 1,







